The term congenital adrenal hyperplasia refers to enlarged adrenal glands. It is due to inherited enzyme deficiency. Congenital adrenal hyperplasia is the most common adrenal disorder of infancy and childhood.
Congenital adrenal hyperplasia results from excessive androgens (male hormones). There is also a severe salt-losing form of the condition.
An enzyme deficiency may result in a failure of the adrenal glands to make normal amounts of cortisol. The decrease in cortisol level stimulates the pituitary gland to release adrenal corticotrophic hormone (ACTH). ACTH makes the adrenal gland enlarge and produce more cortisol, mineralocorticoids and androgens.
Congenital adrenal hyperplasia may be contrasted with Cushings Syndrome, in which there is excess cortisol. When this is due to excess ACTH, the adrenals also enlarge and produce more adrenal androgens.
*Image courtesy Genetics 4 Medics
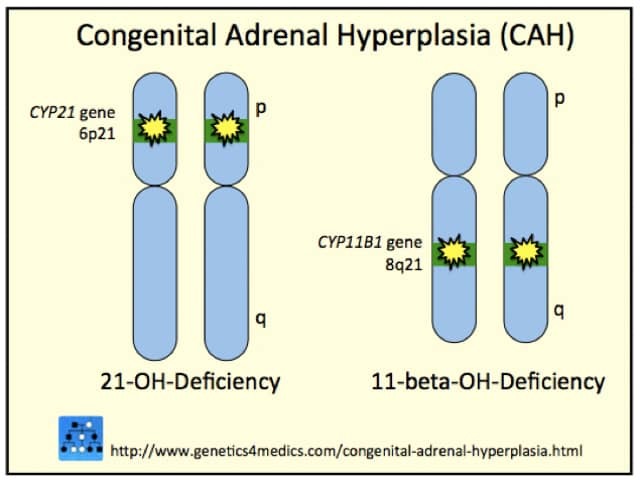
Congenital adrenal hyperplasia may be due to:
The clinical signs of congenital adrenal hyperplasia depend on which enzyme is lacking and to what extent.
21-hydroxylase deficiency or classic congenital hyperplasia accounts for most cases (95%). It may cause health problems in the newborn period or not until puberty or later. The enzyme deficiency reduces cortisol, increases androgens, and in one-third of cases, reduces aldosterone production.
21-hydroxylase deficiency may present in the first two weeks of life. It may present as ‘salt-losing’ acute adrenal insufficiency or as genital ambiguity in females. Partial 21-hydroxylase enzyme deficiency is less severe. It is more likely to present in later childhood or adolescence with signs of androgen excess.
Clinical features of 21-hydroxylase deficiency |
|
|---|---|
Acute adrenal insufficiency |
|
Genital ambiguity |
|
Late-onset in females |
|
Late-onset in males |
|
The diagnosis should be considered in infants presenting with acute adrenal insufficiency, with virilisation (females) or premature puberty (males).
Initial tests in acute adrenal insufficiency reveal a salt-losing state.
21-hydroxylase deficiency is diagnosed by finding high levels of the following hormones in blood or urine:
An adrenal ultrasound scan may be carried out if the baby’s genitals appear abnormal on birth.
Genetic tests may be available to identify the specific genetic mutation.
Genetic testing may be considered during pregnancy if an unborn infant is known to be at risk because a sibling is affected or if both parents are known to carry the abnormal gene. Genetic tests performed during pregnancy include:
Prenatal diagnosis may also be made by finding increased levels of 17-hydroxyprogesterone in amniotic fluid at 14 weeks of gestation.
Treatment of classic congenital hyperplasia aims to replace glucocorticoid (cortisol) to prevent excessive ACTH secretion, using a small dose of dexamethasone, usually 0.5mg at nighttime.
The levels of circulating androgens may be reduced by antiandrogen treatment. Available drugs include:
In the salt-losing form of congenital adrenal hyperplasia, mineralocorticoid, usually fludrocortisone at a dose of 0.1 mg, is given to maintain normal extracellular fluid volume and electrolyte levels. Blood pressure, electrolytes and plasma renin activity are monitored to assess the response.
17-hydroxylase deficiency is rare. It presents at puberty because of reduced adrenal androgen production (hypogonadism).
17-hydroxylase deficiency causes decreased production of cortisol and increased mineralocorticoids. Tests reveal:
Treatment is with dexamethasone to correct hypertension and testosterone to accelerate sexual maturation.
3β-hydroxysteroid dehydrogenase deficiency reduces production of all adrenal steroid hormones (cortisol, aldosterone, androgens and oestrogens). It presents in early infancy with vomiting, salt wasting and genital ambiguity. Tests reveal:
Conversion of pregnenolone to progesterone is impaired, blocking synthesis of both cortisol and aldosterone but increasing adrenal androgens. In females, these weak androgens produce partial virilisation.
3β-hydroxysteroid dehydrogenase deficiency reduces testosterone production in the male testis so that the genitals of a male may be incompletely formed.
Treatment is the replacement of glucocorticoids, fludrocortisone and sex steroids from puberty onwards.

