What is Noonan syndrome?
Noonan syndrome is a common autosomal dominant disorder associated with mutations in the Ras/mitogen–activated protein kinase (MAPK) pathway and is one of a group of conditions that are collectively known as RASopathies.
Noonan syndrome is characterised by:
- A distinctive facial appearance
- Short stature
- Chest deformity
- Congenital heart disease.
Noonan syndrome is also known as:
- Familial Turner syndrome
- Female pseudo-Turner syndrome
- Male Turner syndrome
- Noonan–Ehmke syndrome
- Pseudo–Ullrich–Turner syndrome
- Turner-like syndrome
- Turner phenotype with normal karyotype
- Ullrich–Noonan syndrome.
A 12-year-old girl with Noonan syndrome
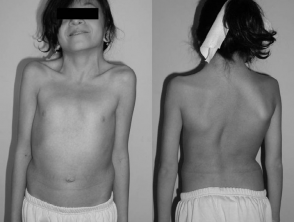
Source: Wikipedia
{kind=link}
What causes Noonan syndrome?
Noonan syndrome is caused by mutations in the genes associated with the Ras/MAPK cell-signalling pathway, which is required for normal cell division, proliferation, differentiation and migration. These mutations lead to the loss of regulation of cell growth and division.
Mutations in the PTPN11 gene are implicated in over 50% of Noonan syndrome cases. Gene mutations in SOS1 are implicated in 10–15% of cases and mutations in the RAF1 gene and the RIT1 gene account for approximately 5% each. A number of other genes account for the rest.
The genetic cause behind Noonan syndrome is unknown in up to 20% of cases.
What are the clinical features of Noonan syndrome?
Noonan syndrome both sexes and all races; 50–70% of people with Noonan syndrome are of short stature. Weight and length at birth are usually normal, but growth slows over time. This is thought to be associated with abnormal levels of growth hormone.
Noonan syndrome presents with distinctive facial features, such as:
- A deep philtrum (groove between the nose and mouth)
- Hypertelorism (widely spaced eyes)
- Low-set ears rotated backwards
- High arched palate
- Poor teeth alignment
- Micrognathia (small lower jaw).
Other clinical manifestations of Noonan syndrome include:
- A webbed neck
- A short neck/low posterior hairline
- Pectus excavatum (sunken sternum) or pectus carinatum (protruding sternum)
- Scoliosis (abnormal lateral curvature of the spine)
- Lymphoedema
- Bleeding disorders
- Hypogonadism (impaired gonad function that can lead to insufficient production of sex hormones).
Congenital heart disease is common in most cases of Noonan syndrome, with the most common defect being pulmonary valve stenosis.
Cutaneous symptoms of Noonan syndrome are varied and literature on the topic is limited. Documented cutaneous effects of Noonan syndrome include:
- Pigmented melanocytic naevi (moles)
- Abnormal dermatoglyphics (fingerprints), with increased numbers of whorls appearing on the fingertips, secondary to peripheral lymphoedema
- Stasis dermatitis (a common inflammatory dermatosis, due to venous pooling in the lower limbs).
- Plantar hyperkeratosis or keratoderma (thickening of the skin on the soles of the feet).
Noonan syndrome with multiple lentigines, formerly known as LEOPARD syndrome, is similar to Noonan syndrome. It has characteristic cutaneous features, including:
- Café-au-lait macules (flat, light brown macules) caused by a collection of pigment-producing melanocytes in the epidermis
- Lentigines (pigmented flat or slightly raised lesions with well-defined borders) — unlike freckles, these spots are not affected by sun exposure.
What are the complications of Noonan syndrome?
Hypertrophic cardiomyopathy is common in individuals with critical congenital pulmonary valve stenosis.
Individuals with Noonan syndrome have an eight-fold increased risk of developing haematological malignancies, the most common being leukaemia.
What is the treatment for Noonan syndrome?
There is no specific treatment for the syndrome. Any complications such as heart disease should be monitored and treated according to symptoms, signs and the results of investigations.