Introduction Demographics Clinical features Diagnosis Treatment
Orofaciodigital syndrome type 1 (OMIM#311200) is a rare genetic disorder that affects females. It is characterised by malformations of the mouth, face and fingers/toes. It is also known as oral-facial-digital syndrome type 1, OFD1 and ‘Papillon-Léage and Psaume syndrome’.
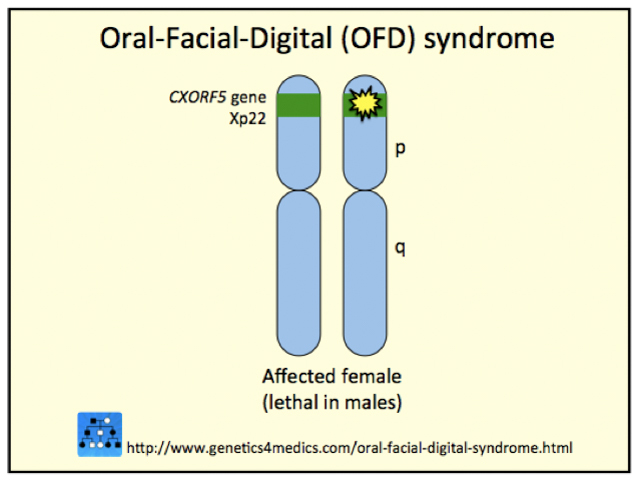
Orofaciodigital syndrome type 1 is an X-linked dominant condition which is lethal for males at the embryo stage. A single copy of the affected gene, which is located on the X chromosome, results in the development of this syndrome. Survival is only possible where there is also one normal copy of the specific gene. As males have only one X chromosome, if the OFD1 gene is abnormal then survival is not possible. Many different mutations of all types have been reported to affect the relevant gene.
OFD1 is classified as a ciliopathy as the gene (now called OFD1, previously CXORF5) is involved in the primary cilia. The primary cilia are key structures in cell function and are short, microscopic, hairlike vibrating structures on the cell surface.
OFD1 is reported to affect 1/250,000 live births. A negative family history for OFD1 is found in 75% of cases.
The classic features of OFD1 involve the mouth, face and digits (fingers/toes). There is considerable variability in clinical presentation between affected families and even within families.
Mouth |
|
|
Face |
-Widely spaced eyes
|
|
Digits |
|
|
Skin |
|
|
Hair |
|
|
Kidney |
|
|
Nervous system |
|
|
Other organs |
|
|
*Image courtesy Genetics 4 Medics
OFD1 is suspected clinically in a baby with the typical mouth, facial and finger/toe anomalies. It is distinguished from the other orofaciodigital syndromes on the inheritance pattern and polycystic kidneys. Milial cysts and hair loss have also only been reported in OFD1. Diagnosis can be difficult in patients with only minor features.
The gene mutation can be identified by DNA sequencing in specialist research laboratories.
Ultrasound of the kidneys and monitoring of kidney function is recommended.
There is no specific treatment for this syndrome. Treatment is directed at the problems encountered in an individual. For some patients the kidney problems dominate the clinical course, resulting in kidney failure and kidney transplant.

