What is IgG4?
IgG4 is a subclass of IgG, which is the most common form of immunoglobulin. IgG accounts for 75% of antibodies circulating in the blood, which are an essential part of the secondary immune response to infection and toxins. IgG is made by plasma cells, a specific type of B lymphocyte.
There are four subclasses of IgG. Subclass IgG4 is the least common of these, accounting for about 4% of IgG in serum. IgG4 has a unique structure. Its specific biological role is uncertain. However, IgG4 is known to play a role in protection against type 1 hypersensitivity reactions, for example to bee venom, and in the pathogenesis of autoimmune blistering diseases such as pemphigus vulgaris, helminth infections, and malignancy including melanoma.
What is IgG4-related disease?
IgG4-related disease is a newly-described rare syndrome consisting of many disease entities that were previously thought to be unrelated. These conditions have the pathological features of:
- Inflammatory pseudotumours, that is, organomegaly or nodules within an organ
- Lymphoplasmacytic infiltration of tissues with many IgG4-positive plasma cells
- Storiform fibrosis (scarring involving cells arranged like a cartwheel on histology).
IgG4-related disease: cutaneous inflammatory pseudotumour
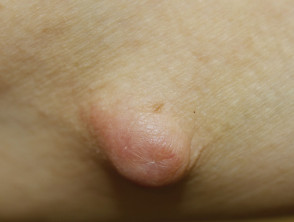
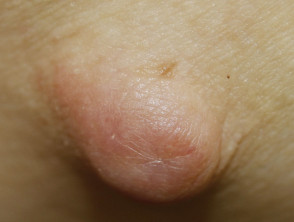
Elevated serum IgG4 is present in 60–70% patients.
What is the cause of IgG4-related disease?
The cause of IgG4-related disease is unknown.
- It may be an autoimmune disorder: antinuclear antibodies and autoantibodies to pancreatic antigens have been found in autoimmune pancreatitis.
- It has features of an allergic disorder: abnormally high Th2 cytokines in tissues, raised IgE and increased T-reg lymphocytes in the blood, and raised eosinophil count in 40% of patients. Cytokines IL-10 (interleukin 10) and TGF-β (transforming growth factor Beta) are known to support IgG4 production and are at elevated levels in IgG4-related disease.
Who gets IgG4-related disease?
Most patients described with IgG4-related disease are middle-aged or older men.
What are the clinical features of IgG4-related disease?
IgG4-related disease presents in various ways.
- Most people with IgG4-related disease are constitutionally well.
- The IgG4-related disease may be diagnosed as an incidental finding when someone undergoes a routine radiological or surgical investigation.
- 60–90% of people with IgG4-related disease have multiple organ involvement.
- The IgG4-related disease often presents as swelling of or within an organ (such as skin, orbit and lung); this is known as an inflammatory pseudotumour.
- IgG-related autoimmune pancreatitis is more likely than other forms of IgG-related disease to be associated with disease in other sites, such as hilar lymphadenopathy, bile duct lesions, lacrimal and salivary gland involvement, hypothyroidism, and retroperitoneal fibrosis.
- IgG4-related lacrimal, parotid, or submandibular salivary disease, pneumonitis, and kidney disease are associated with pancreatitis in < 20% cases.
- Patients with IgG4-related disease may have symptoms of allergy such as asthma.
- Up to 40–80% of patients with IgG4-related disease have lymphadenopathy.
- Cutaneous manifestations of IgG4-related disease include erythematous plaques, papules, and subcutaneous nodules, often itchy, and usually on the face or forearm. Most patients with cutaneous IgG4-related disease report inflammatory enlargement of lacrimal or salivary glands at some point.
| Organ | Disease |
|---|---|
| Gastrointestinal disease |
|
| Lymph node involvement |
|
| Salivary and lacrimal disease |
|
| Eye disease |
|
| Heart disease |
|
| Thyroid disease |
|
| Lung disease |
|
| Kidney disease |
|
| Skin disease |
|
| Other |
|
How is IgG4-related disease diagnosed?
The diagnosis of IgG4-related disease can be difficult, as multiple organs may be involved simultaneously. Diagnostic criteria have not been fully developed.
Investigation in suspected IgG4-related disease requires a combination of clinical, endoscopic, radiological and serological tests looking for organ involvement and end-organ damage (eg, hormonal abnormalities).
Tissue diagnosis requires a biopsy of affected organ tissues, including skin biopsy. See IgG4-related skin disease pathology. Features include:
- Lymphoplasmacytic infiltrate
- Storiform fibrosis pattern
- IgG4-positive plasma cells (the number of positive cells to confirm diagnosis depends on tissue type; most > 30–50 per high power field, some, such as kidney, 10 per high power field).
Blood tests are not diagnostic but may show:
- Peripheral eosinophilia
- Raised serum IgG4.
There are specific criteria for the diagnosis of some tissue-specific disorders, such as autoimmune pancreatitis.
How is IgG4-related disease treated?
IgG4-related disease is usually treated with systemic steroids, often prednisone 40 mg per day for 2–4 weeks followed by a gradual tapering of the dose.
In patients that cannot be taken off prednisone, a steroid-sparing agent like azathioprine or mycophenolate may be used. Rituximab, a B cell-depleting monoclonal antibody, has been used with some success.
Organ-specific replacement therapy may be required:
- Thyroxine for thyroiditis (thyroid disease) causing hypothyroidism
- Pancreatic enzyme replacement for pancreatic insufficiency
- Insulin for diabetes mellitus
- Hormone replacement for hypopituitarism: hydrocortisone, thyroxine, growth hormone, desmopressin and sex hormones (testosterone for men; oestrogen and progesterone for women).
What is the prognosis of IgG4-related disease?
Prognosis of IgG4-related disease is variable. It may spontaneously resolve or persist, with remitting and relapsing symptoms.
Major causes for morbidity and mortality are significant organ involvement such as:
- Liver cirrhosis
- Portal hypertension
- Biliary obstruction
- Retroperitoneal fibrosis
- Aortic dissection from an aneurysm
- Diabetes mellitus
- Pancreatic insufficiency
IgG4-related disease may be associated with a possible increased risk of non-Hodgkin lymphoma. It is not known if IgG4-related disease leads to an increased risk of other forms of cancer.