Angelman syndrome is a rare neurological disorder which occurs in 1 out of every 15,000 births and in the past, was mistaken for other disorders like cerebral palsy or autism. It is marked by a complex array of symptoms.
It was named for Dr. Harry Angelman, who first described the disorder in 1965.
Angelman syndrome is rare:
Angelman syndrome is genetic in origin. Genetic changes can be random, that is, without a family history of the disorder.
Prader-Willi syndrome is a clinically distinct disorder due to a paternally derived defect mapped to the same chromosome as Angelman syndrome.
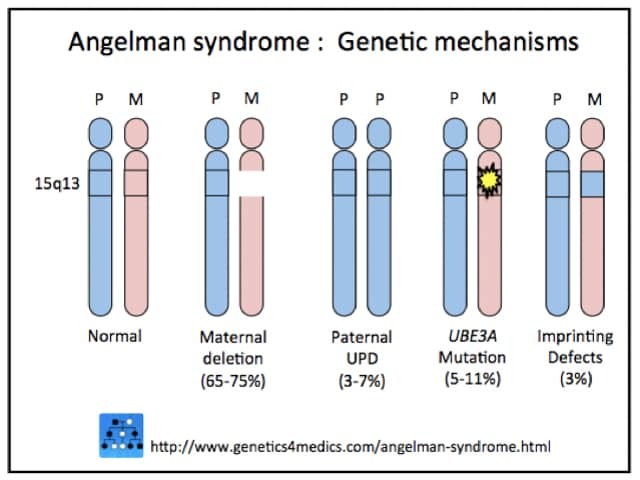
*Image courtesy Genetics 4 Medics
Cutaneous features of Angelman syndrome include:
Noncutaneous features of Angelman syndrome include seizures, developmental delays, limited or lack of speech, mobility disorders, increase in smiling, a happy, excitable personality, hand flapping, abnormal sleep cycles and microcephaly.
There is no cure for Angelman syndrome. Lifelong care is needed and treatment focuses on managing symptoms. It can include:
Despite the many limitations, the life expectancy of patients with Angelman syndrome is normal. Often, patients will become less excitable as they age and they outgrow sleep cycle abnormalities.

