Introduction Introduction - MEN type 2B disease Causes Diagnosis Treatment
Multiple endocrine neoplasia (MEN) syndromes are mostly inherited conditions in which several endocrine glands develop benign (non-cancerous) or malignant (cancerous) tumours or hyperplasia (grow excessively without forming tumours).
There are 2 main types of MEN, MEN type 1 and MEN type 2. MEN type 2 is further split into 3 subtypes:
Multiple endocrine neoplasia type 2B disease has additional features including mucosal neuromas (nerve tumours on the mucous membranes), neuromas in the gut that lead to gastrointestinal abnormalities, and striking facial appearance associated with Marfanoid habitus (slender, tall, long fingers and toes, and high arched palate like Marfan disease). These features may be the first sign of an internal malignancy and should prompt further investigation.
Like MEN type 2A disease, MEN type 2B disease carries a high risk for development of medullary carcinoma of the thyroid and phaeochromocytoma (a vascular tumour of the adrenal gland that may cause high blood pressure).
MEN type 2B disease is also characterised by the development in early life of multiple mucosal neuromas. Neuromas appear as:
Patients with MEN type 2B disease also often develop spinal abnormalities, and abnormalities of the bones in the feet and thighs. Many have long limbs and loose joints. These features, along with thickened lips and eyelids are associated with Marfanoid habitus (the features of Marfan syndrome).
In 95% of cases MEN type 2B disease is due to a mutation in the tyrosine kinase domain of the RET gene at codon 918 in exon 16. The RET gene is a protocogene which means that a mutation can predispose to the formation of cancers.
All MEN 2 subtypes are inherited in an autosomal dominant manner and offspring of affected individuals have a 50% chance of inheriting the disease. However, not all MEN type 2B cases are inherited, as there are some people with the disease whom have no family history of it. It has been found that about 50% of affected individuals inherit the mutation from a parent, and 50% have a new mutation.
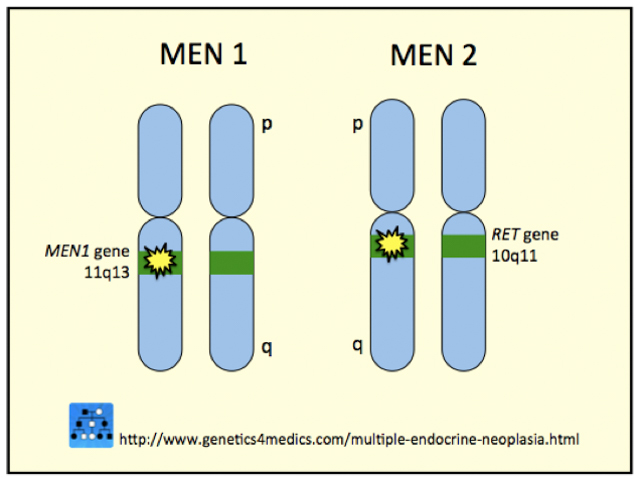
*Image courtesy Genetics 4 Medics
Diagnosis of MEN type 2B is made clinically by the presence of:
Molecular genetic testing can be used to confirm the diagnosis, for predictive testing and for prenatal diagnosis.
Individuals with MEN type 2B disease should have their thyroid gland removed at a very early age (around 1 year) to reduce the risk of thyroid cancer. Compared with other types of thyroid cancer, medullary carcinoma of the thyroid is a very aggressive cancer. If the thyroid is not removed in childhood, the average age of death in people with MEN type 2B disease is around 21 years. Once the thyroid is removed, patients must take thyroid hormone replacements (thyroxine) for the rest of their life.
There is no known cure for any of the MEN syndromes. Patients are treated for their symptoms. Annual urine biochemical screening for phaeochromocytoma is recommended for all patients. Genetic testing is also important for people with a family history of the disease as early diagnosis and treatment will reduce the risk of thyroid cancer and death.

