

Subtypes of Ehlers–Danlos syndrome — extra information
The 2017 international classification of Ehlers-Danlos syndromes
The 2017 international classification of Ehlers–Danlos syndromes
Specific subtypes of Ehlers–Danlos syndrome (EDS), or Ehlers–Danlos syndromes, are described below [1,2]. They include:
- Classical Ehlers–Danlos syndrome (cEDS)
- Classical-like Ehlers–Danlos syndrome (clEDS)
- Cardiac-valvular Ehlers–Danlos syndrome (cvEDS)
- Vascular Ehlers–Danlos syndrome (vEDS)
- Hypermobile Ehlers–Danlos syndrome (hEDS)
- Arthrochalasia Ehlers–Danlos syndrome (aEDS)
- Dermatosparaxis Ehlers–Danlos syndrome (dEDS)
- Kyphoscoliotic Ehlers–Danlos syndrome (kEDS)
- Brittle cornea syndrome (BCS)
- Spondylodysplastic Ehlers–Danlos syndrome (spEDS)
- Musculocontractural Ehlers–Danlos syndrome (mcEDS)
- Myopathic Ehlers–Danlos syndrome (mEDS)
- Periodontal Ehlers–Danlos syndrome (pEDS)
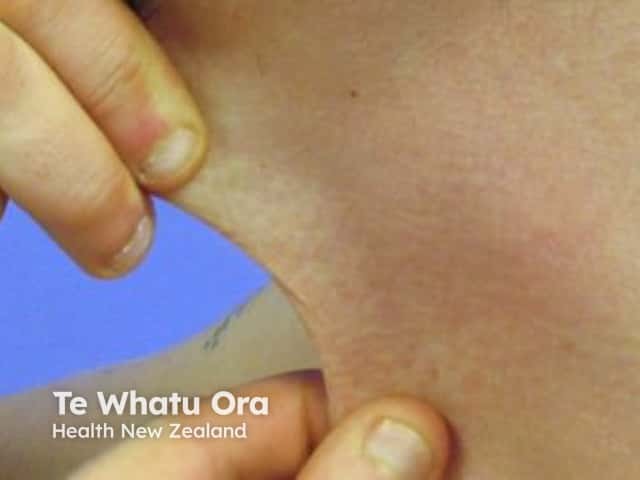
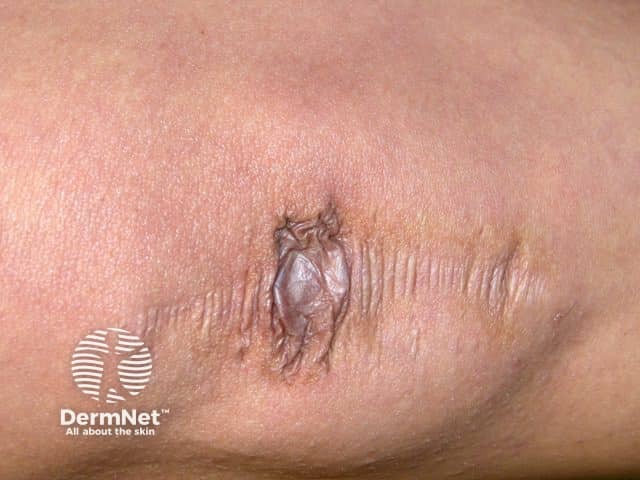
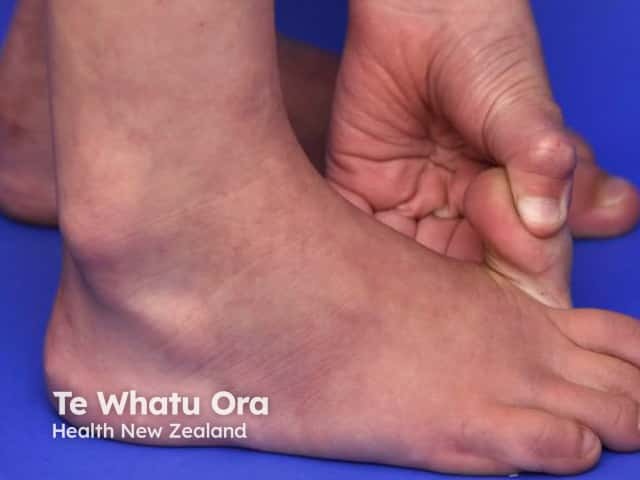
See more images of Ehlers–Danlos syndrome.
Classical Ehlers–Danlos syndrome (cEDS)
The inheritance pattern of cEDS is autosomal dominant.
- The affected genes in cEDS are COL5A1 (MIM 130000), COL5A2, (MIM 130010), and COL1A1 affecting Type V and Type I collagen.
- 50% of patients diagnosed with cEDS have a de-novo mutation (ie, neither parent is affected).
- The severity varies significantly, even within the same family.
- Vascular fragility may include spontaneous dissection or rupture of medium-sized arteries.
- Typically, patients with cEDS have marked skin hyperextensibility, widened atrophic cutaneous scars, and generalised joint hypermobility.
- Other features of cEDS are haemosiderin deposition (particularly noticeable on shins and backs of hands), subcutaneous spheroids (calcified fat lobules after losing their blood supply), and molluscoid pseudotumours (thickened fleshy skin lesions/scars over the knees and elbows).
- There are no striae (stretch marks) in cEDS.
- Typical electron microscopy findings are 'collagen flowers' or 'cauliflowers' due to abnormal fibrillogenesis.
- Complications of cEDS are heart valve deformities, aorta dilatation, and aortic dissection
Classical-like Ehlers–Danlos syndrome (clEDS)
The inheritance pattern of clEDS is autosomal recessive.
- The affected gene in clEDS is TNXB affecting the protein Tenascin XB (MIM 606408)
- Patients with clEDS have generalised joint hypermobility, hyperextensible, soft and velvety skin, and severe bruising.
- There is no atrophic scarring in clEDS.
Cardiac-valvular Ehlers–Danlos syndrome (cvEDS)
The inheritance pattern of cvEDS is autosomal recessive.
- The affected gene in cvEDS is COL1A2 affecting the protein Type I collagen (MIM 225320)
- Patients with cvEDS present with minor signs of EDS but have severe aortic defect/cardiac valve involvement requiring surgical intervention.
Vascular Ehlers–Danlos syndrome (vEDS)
The inheritance pattern of vEDS is autosomal dominant.
- The affected genes in vEDS are COL3A1 affecting Type III collagen and COL1A1 affecting Type 1 collagen (MIM 130050)
- 50% of patients diagnosed with vEDS have a de novo mutation (ie, neither parent is affected).
- The median life expectancy is 50 years.
- The subtle or absent clinical features of vEDS in some patients make this subtype difficult to diagnose, with sudden death in the ages 30s or 40s being the only presenting feature.
- vEDS can be identified at birth if there are noticeable clubfoot deformity or hip dislocation.
- Features of vEDS are severe bruising, thin translucent skin, and characteristic facial appearance (taut and hollow cheeks, thin lips and nose, and prominent eyes).
- Complications of vEDS are arterial dissection (including aorta), rupture (including bowel rupture, with sigmoid colon the commonest site), and obstetric complications (uterine rupture, massive postpartum haemorrhage).
Hypermobile Ehlers–Danlos syndrome (hEDS)
The inheritance pattern of hEDS is autosomal dominant.
- The affected gene in hEDS is unknown (MIM 130020).
- Patients with hEDS are not prone to life-threatening complications, but its effects can be debilitating.
- Features of hEDS are generalised joint hypermobility, spine hypermobility, and frequent joint dislocations or subluxations (shoulder, patella or temporomandibular joint).
- There are no molluscum pseudotumours.
- Complications of hEDS are aortic root dilatation, autonomic dysfunction (postural orthostatic tachycardia syndrome), severe chronic joint pain, scoliosis, and premature osteoarthritis.
Arthrochalasia Ehlers–Danlos syndrome (aEDS)
The inheritance pattern of aEDS is autosomal dominant.
- The affected genes in aEDS are COL1A1 and COL1A2 affecting Type I collagen (MIM 130060).
- As the word describes, arthro- means joint, and -chalasia means relaxation; the mutation results in abnormal weak collagen.
- Patients with aEDS have a lifelong risk for concurrent dislocation of multiple major joints.
- Features of aEDS are hypotonia, severe generalised joint hypermobility, congenital bilateral hip dislocation, and osteopenia.
Dermatosparaxis Ehlers–Danlos syndrome (dEDS)
The inheritance pattern of dEDS is autosomal recessive.
- The affected gene in dEDS is ADAMTS2 affecting the protein ADAMTS-2 (MIM 225410).
- Sparaxis is derived from a Greek word which means 'to tear'.
- Features of dEDS are severe skin fragility, lax redundant skin, delayed closure of the fontanelles, blue sclerae, visceral fragility, and post-natal growth retardation resulting in short stature.
- There are characteristic 'hieroglyphic fibrils' on electron microscopy of the skin.
Kyphoscoliotic Ehlers–Danlos syndrome (kEDS)
The inheritance pattern of kEDS is autosomal recessive.
- The affected genes in dEDS are PLOD1 affecting the protein LH1 (MIM 225400), and FKBP14 affecting the protein FKBP22 (MIM 614557).
- Gene mutations result in a deficiency of lysylhydroxylase, which is a collagen-modifying enzyme important for forming crosslinks between collagen trimers to increase collagen strength.
- kEDS is characterised by early-onset progressive kyphoscoliosis, hypotonia, gross motor delay, blue sclerae with raised intraocular pressure, generalised joint hypermobility, and easy bruising.
Brittle cornea syndrome (BCS)
The inheritance pattern of BCS is autosomal recessive.
- The affected genes in dEDS are PRDM5 affecting the protein PRDM5 (MIM 614170) and ZNF469 affecting ZNF469 (MIM 229200).
- Patients with BCS have a thin and fragile cornea that can perforate spontaneously or after minor trauma.
- The features of BCS are thin cornea, keratoconus, blue sclerae, risk of globe rupture, retinal detachment, and high myopia (nearsightedness).
Spondylodysplastic Ehlers–Danlos syndrome (spEDS)
The inheritance pattern of spEDS is autosomal recessive.
- The affected genes in spEDS are B4GALT7 affecting the protein beta4GalT7 (MIM 130070), B3GALT6 affecting beta3GalT6 (MIM 615349), and SLC39A13 affecting ZIP13 (MIM 612350).
- The features of spEDS are short stature, hypotonia, limb-bowing, osteopenia, and hyperextensible, thin, doughy skin.
Musculocontractural Ehlers–Danlos syndrome (mcEDS)
The inheritance pattern of mcEDS is autosomal recessive.
- The affected genes in mcEDS are CHST14 affecting the protein D4ST1 (MIM 601776) and DSE affecting DSE (MIM 615539).
- The features of mcEDS are congenital contractures resulting in club foot, recurrent dislocations, adducted (clasped) thumbs in infancy, hyperextensible skin, easy bruising, fragile skin, and atrophic scars.
Myopathic Ehlers–Danlos syndrome (mEDS)
The inheritance patterns of mEDS can be autosomal dominant or autosomal recessive.
- The affected gene in mEDS is COL12A1 affecting Type XII collagen (MIM 616471).
- Muscle weakness present in infancy and childhood tends to improve until young adulthood, with some deterioration after the age of about 40.
Periodontal Ehlers–Danlos syndrome (pEDS)
The inheritance pattern of pEDS is autosomal dominant.
- The affected genes in pEDS are C1R affecting the protein C1r and C1S affecting C1s (MIM 130080).
- The features of pEDS are early-onset severe periodontitis (swollen red gums, gum recession, early loss of teeth), yellow-brown bruise-like pretibial plaques, joint hypermobility, and hyperextensible skin.
References
- Malfait F, Francomano C, Byers P, et al. The 2017 International Classification of the Ehlers-Danlos Syndromes. Am J Med Genet Part C Semin Med Genet. 2017 Mar;175C(1):8–26. doi: 10.1002/ajmg.c.31552. Journal (PDF)
- Brady AF, Demirdas S, Fournel-Gigleux S, et al. The Ehlers-Danlos syndromes, rare types. Am J Med Genet Part C Semin Med Genet. 2017 Mar;175C(1):70–115. doi: 10.1002/ajmg.c.31550. Journal
On DermNet
Other websites
- Ehlers Danlos syndromes — National Organization for Rare Disorders
- What are the Ehlers-Danlos syndromes? — Ehlers-Danlos Support, UK