Introduction
Demographics
Subtypes
Causes
Clinical features
Complications
Diagnosis
Differential diagnoses
Treatment
Outcome
Ehlers–Danlos syndrome (EDS) is a group of inherited disorders that involve a genetic defect in collagen or connective tissue synthesis and structure. This results in:
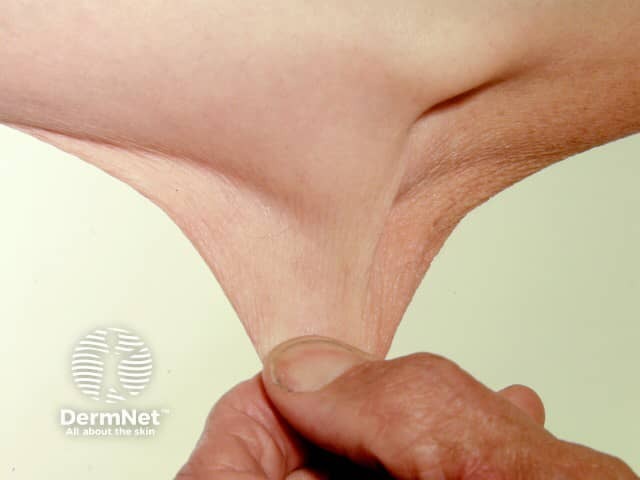
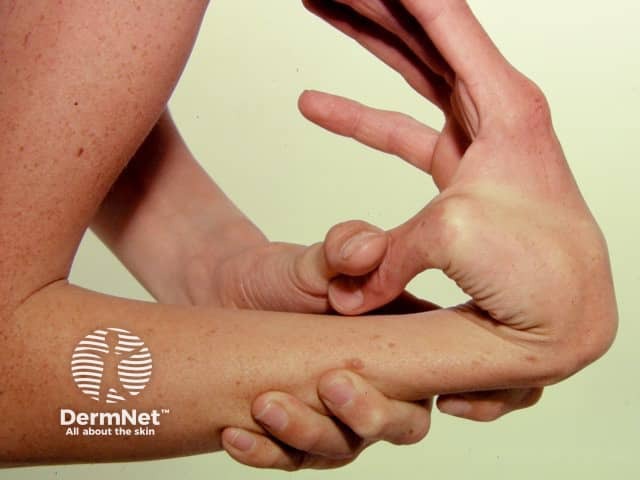
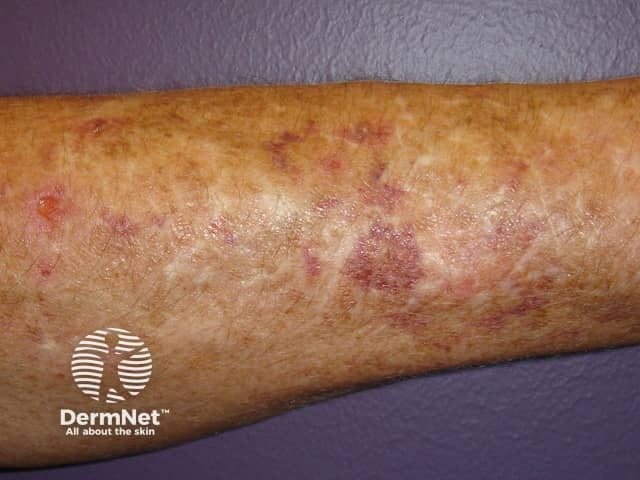
The global frequency of EDS is about 1 in 5000. EDS may occur in males and females of all races and usually first becomes apparent in childhood and young adult life.
EDS has autosomal dominant and autosomal recessive inheritance patterns.
The 2017 International Classification for Ehlers-Danlos Syndromes, describes thirteen subtypes of EDS, classified according to the clinical features, inheritance pattern, and molecular genetic defects.
Each is a distinct disorder that ‘runs true’ in a family. This means that members of a family affected by EDS will share the same features. Some cases do not fit neatly into a known type of EDS, and a patient may show features of more than one type. Clinical manifestations and their severity can vary significantly, even within the same family.
Hypermobile EDS is the most common subtype of EDS (MIM 130020), followed by classical EDS (MIM 130000) and vascular EDS (MIM 130050). The other types of EDS are very rare (less than 10%). A 14th subtype was identified in 2018.
See Subtypes of Ehlers–Danlos syndrome
Collagen is one of the main building blocks of the body. It is a protein that is widely found in the structure of many tissues and organs of the body. Several types of collagen exist, each with differing properties. Collagen can provide strength and firm support, it can be elastic to allow movement, or it can be used to bind things together.
A genetic defect can cause reduced amounts of collagen, collagen disorganisation (collagen is usually organised into bundles), and alterations in the size and shape of collagen. The subtype of EDS depends on how collagen metabolism has been affected. For example, vascular EDS is caused by decreased or absent synthesis of type III collagen.
There are some types of EDS that are a result of other extracellular matrix disorders and its components (such as glycosaminoglycans) and of defects in intracellular processing.
Signs and symptoms differ in type and severity between the different subtypes of EDS, and can be quite specific to a subtype. The characteristic clinical features of EDS are hyperextensible skin, skin fragility, easy bruising, and joint laxity.
Classic Ehlers-Danlos syndrome (MIM 130000) has the additional cutaneous features:

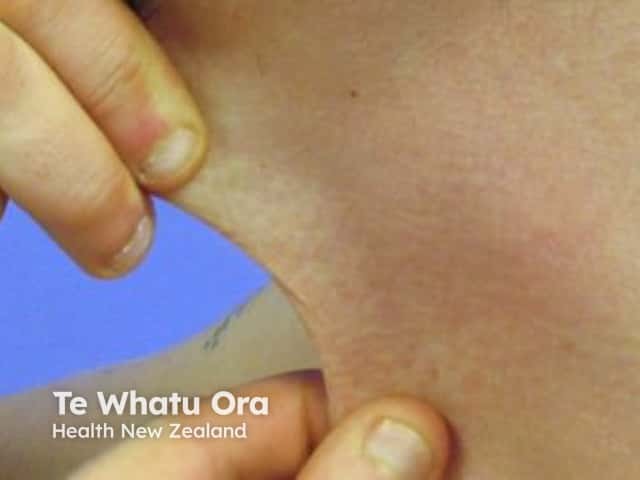
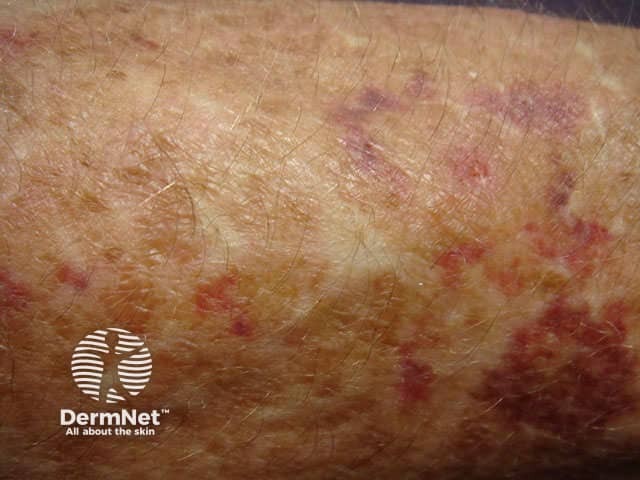
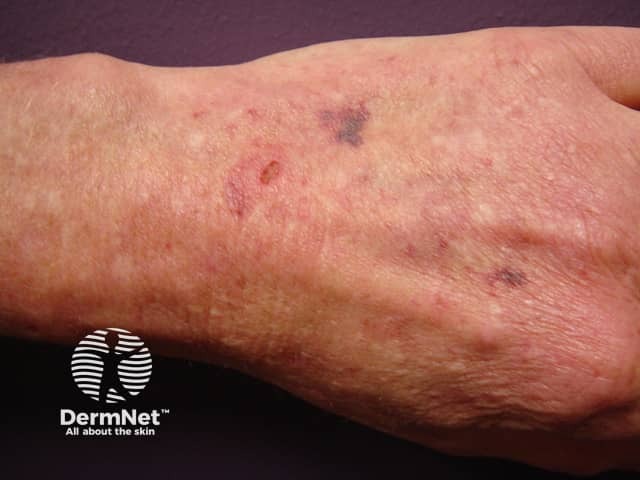
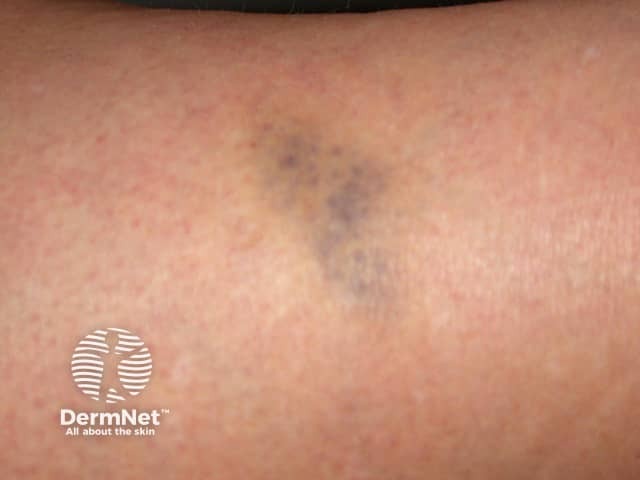
Other forms of EDS may show:
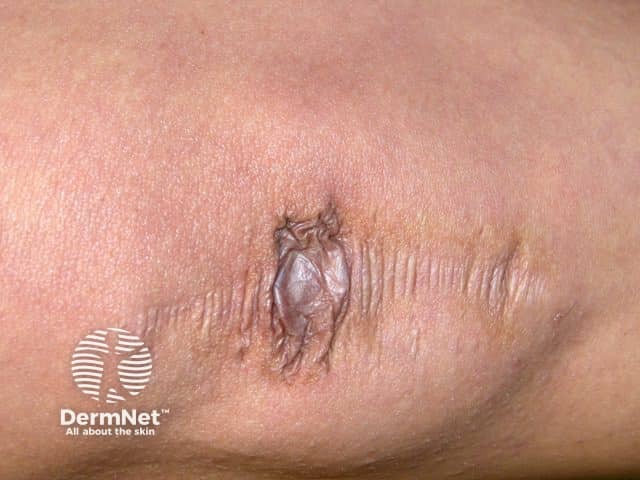
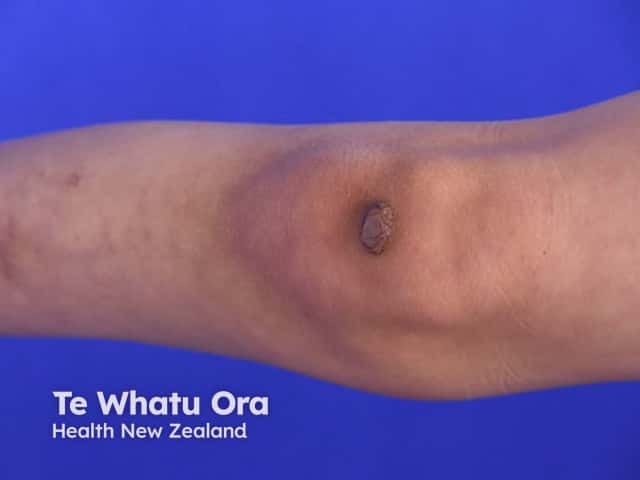
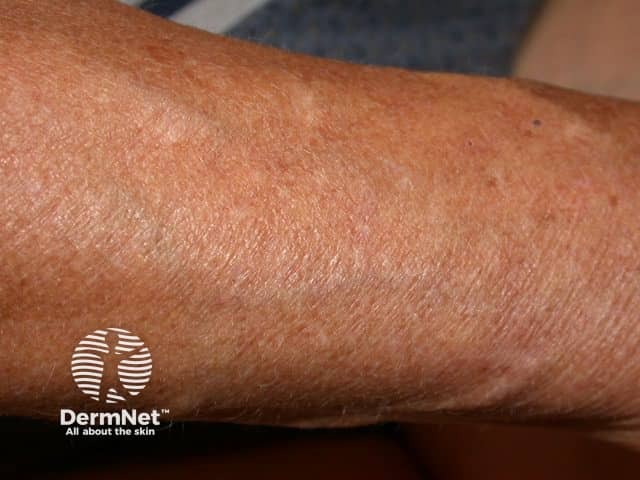
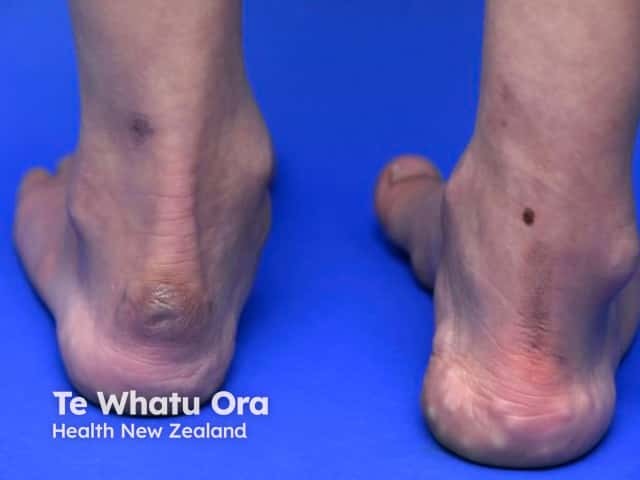
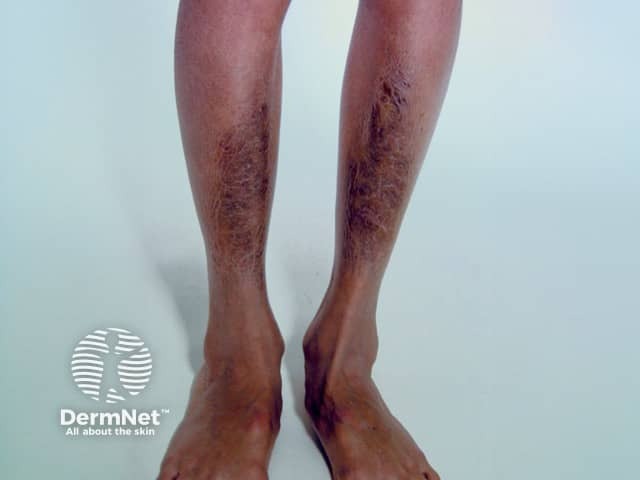
Symptoms and signs can be quite specific to an EDS subtype.

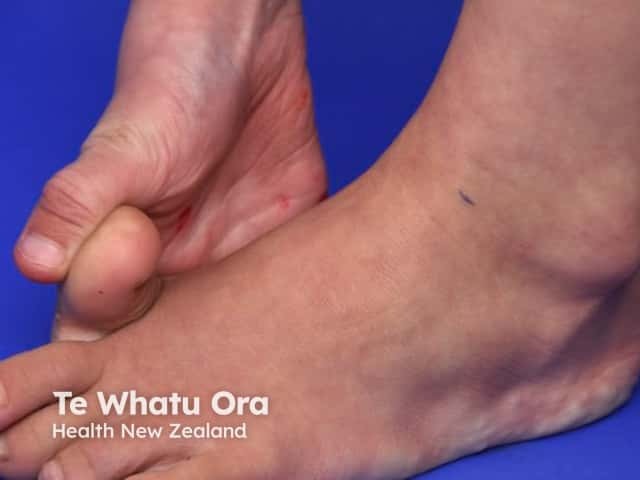
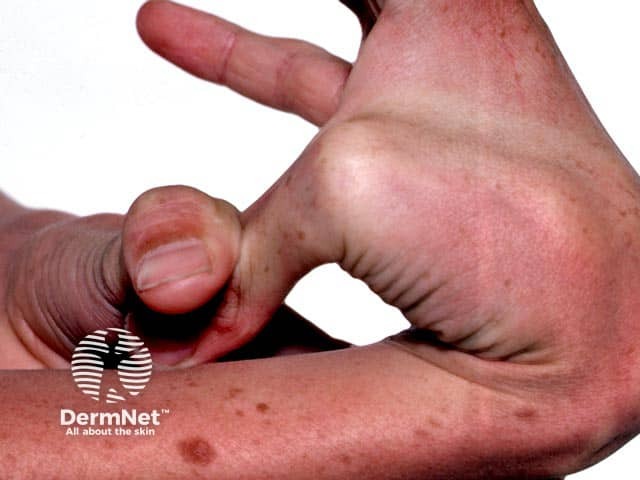
See more images of Ehlers–Danlos syndrome.
Possible complications of Ehlers-Danlos syndrome include:
Suspicion of Ehlers-Danlos syndrome should lead to a careful history and examination. Major and minor criteria have been developed for each of the 13 subtypes of EDS. The definite diagnosis of a specific variant requires tissue biopsy and molecular genetic analysis due to the variety of molecular defects and clinical overlap between EDS subtypes.
Detailed personal and family history of muscle, ocular, cutaneous, dental, cardiac, and respiratory conditions.
Molecular genetic testing confirms the diagnosis, except in the case of hypermobile EDS, which has an unknown genetic basis and so is a clinical diagnosis which must meet all the clinical criteria.
Management guidelines depend on the subtype of EDS.
The prognosis of patients with Ehlers-Danlos syndrome varies widely even within the same subtype. The majority of patients have a normal life expectancy and cognitive function. Individuals with vascular EDS may have a shorter life expectancy and are at higher risk of sudden death due to internal organ and major vessel rupture.

