

Wilson disease — extra information
What is Wilson disease?
Wilson disease is a rare inherited disorder that results in excessive amounts of copper in the body. It is four times more common in females than in males. Copper is normally metabolised by being incorporated into copper-containing enzymes called ceruloplasmin and being excreted into the bile. However, in Wilson disease, the process is impaired and excess copper is initially deposited in the liver where it damages liver cells. Eventually, as liver copper levels increase, it is released into the blood and deposited in other organs, particularly the brain and spinal cord. The copper deposits cause tissue damage, tissue death, and scarring, which cause the affected organs to stop functioning properly. Wilson disease is also known as hepatolenticular degeneration.
What are the signs and symptoms of Wilson disease?
Although abnormal copper accumulation is happening from birth, the symptoms of Wilson disease may not become apparent until late childhood or adolescence. Because copper initially accumulates in the liver, the first sign of the disease is usually liver-related and manifests as one of the following:
- Hepatitis (acute or chronic inflammation of the liver)
- Cirrhosis (severe liver disease due to a progressive loss of liver function)
- Fulminant hepatic failure (sudden and often fatal liver disease)
The most common initial presentation is with cirrhosis, usually occurring during the first 10 years of life. Possible symptoms may include:
- Confusion and drowsiness (hepatic encephalopathy)
- Bleeding from the intestines due to high pressure in compressed veins within the liver (portal hypertension and oesophageal varices)
- Fluid accumulation in the abdomen (ascites) and legs (oedema)
- Susceptibility to infections
- Excess fluid in the lungs causing breathlessness
Other symptoms that may occur as copper is deposited into other organs include:
Neurological symptoms – usually becoming apparent around the 2nd or 3rd decade of life, but may even occur as late as 50 years of age
- Tremor of the head, arms, or legs
- Impaired muscle tone causing unpredictable, jerky, repetitive, or slow movements
- Slurred or slow speech, excessive saliva, decreased or slow facial expressions (mask-like facies)
- Emotional or behavioural changes
- Confusion, delirium or dementia
Eye symptoms
- Kayser-Fleischer rings (coloured rings) formed by deposits of copper in the corneal membrane. Colour of the rings range from greenish-gold to brown.
Muscles and joints
- Stiffness and difficulty moving extremities as joints become involved
- Softening and thinning of the bones
Skin changes
- Yellowish discolouration of skin and eyes (jaundice)
- Development of unusually dark skin patches (hypermelanotic pigmentation)
- Other signs of liver disease: spider telangiectases and palmar erythema (red palms)
Blood problems
- Haemolytic anaemia
- Thrombocytopenia (low levels of platelets) resulting in easy bruising and bleeding because of defective clotting
Kidney disease
- Symptoms are variable
- Kidney or bladder stones may occur
- Blood may be seen in the urine (haematuria)
What is the cause of Wilson disease?
Wilson disease is caused by mutations to the ATP7B gene located on the long arm (q) of chromosome 13. The protein (copper-transporting adenosine triphosphatase) regulated by this gene plays a role in the transport of copper. The defective gene in Wilson disease is inherited in an autosomal recessive manner. For an individual to have Wilson disease they must have inherited a defective gene from each parent. A person who has one defective gene and one normal gene is termed a “carrier” and will lead a perfectly healthy life. However, the carrier is able to pass on the defective gene to a son or daughter.
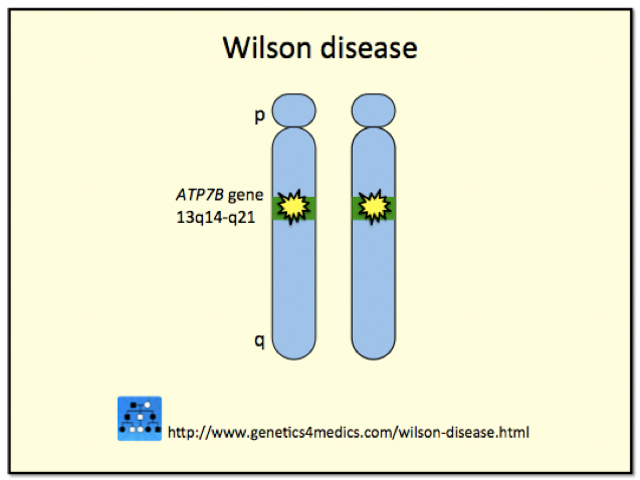
*Image courtesy of Genetics 4 Medics
How is the diagnosis of Wilson disease made?
Several tests can be done to confirm the diagnosis of Wilson disease. The presence of Kayser-Fleischer rings and ceruloplasmin levels of less than 20 mg/dL, in a patient with neurological signs and symptoms suggest the diagnosis of Wilson disease.
Laboratory finding may include:
- Abnormal liver function tests: elevated bilirubin and transaminase enzyme levels and low amounts of circulating albumin
- Low serum ceruloplasmin (in 5-10% of patients with Wilson disease levels may be normal)
- Serum copper levels of less than 80 mcg/dL (in spite of copper deposits in tissue)
- Urine copper levels of greater than 100 mg
- Low serum uric acid levels
- Tissue sample through surgical biopsy of the liver show increased levels of copper (greater than 250 mcg/g dry weight)
Physical examination may show signs of liver or spleen disorders, including cirrhosis and liver necrosis. X-ray examinations including MRI or head CT scan may show abnormalities, especially around the basal ganglia in the brain. Abdominal scans may indicate liver disease or other abnormalities.
Genetic testing for Wilson disease has not yet been developed.
What is the treatment for Wilson disease?
The goals of treatment are to reduce the amount of copper in the tissues and to manage the symptoms and complications of the disorder.
To remove excess copper from the body and prevent ongoing copper accumulation, chelating agents and medications that block copper absorption from the gastrointestinal tract are used.
- Zinc acetate – blocks the absorption of copper in the GI tract
- Penicillamine – forms soluble complexes with copper which is excreted in urine
- Trientine – similar action to penicillamine and is a suitable alternative for people who cannot tolerate penicillamine. Should be given in conjunction with zinc.
A low copper diet is also recommended. Avoid eating foods with a high copper content such as liver, mushrooms, chocolate, nuts, dried fruit, and shellfish.
Patients with Wilson disease will require lifelong treatment to control the disorder. Wilson disease may be fatal because of loss of liver function and toxic effects of copper on the nervous system, hence patients need to be monitored regularly to address early, and avoid complications.
Patients with severe liver disease may be offered liver transplantation.
References
- OMIM – Online Mendelian Inheritance in Man (search term Wilson disease)
- Wilson Disease — GeneTests GeneReviews
- Book: Textbook of Dermatology. Ed Rook A, Wilkinson DS, Ebling FJB, Champion RH, Burton JL. Fourth edition. Blackwell Scientific Publications.
On DermNet
Other websites
- Genetics of Wilson Disease – Medscape Reference
- Wilson Disease — emedicine dermatology, the online textbook
- Wilson disease UK support group
- Wilson Disease Association