Alkaptonuria is a rare genetic disease that is characterised by passing urine that becomes black when left standing. The earliest sign of the condition is usually dark staining found in nappies or diapers of infants.
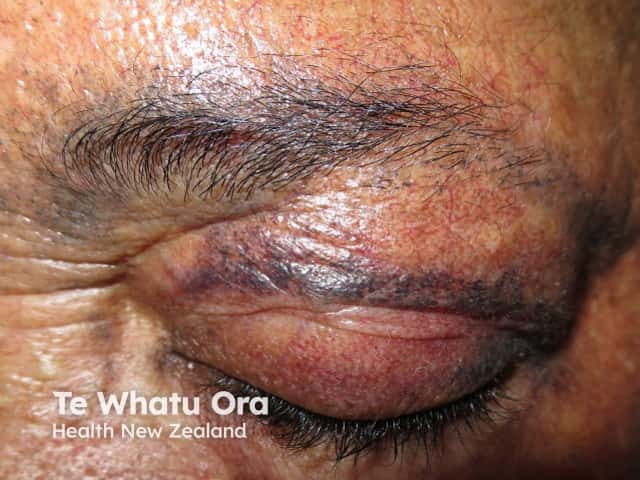
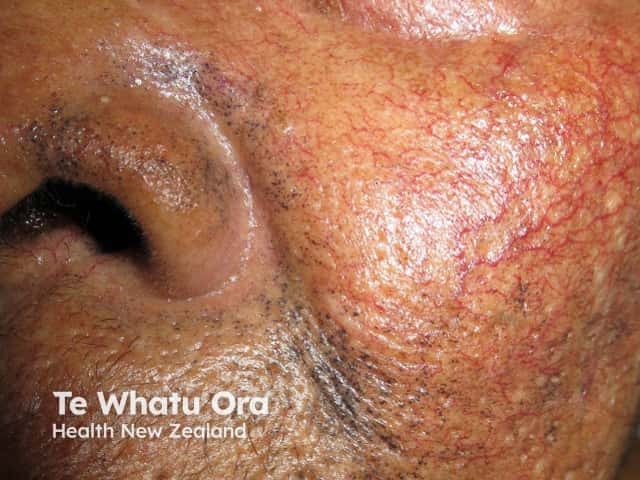
The most obvious sign in adults is a thickening and blue-black discolouration of the ear cartilage. This blue-black discolouration of connective tissue (including bone, cartilage, and skin) caused by deposits of yellow or ochre-coloured pigment is called ochronosis.
Alkaptonuria is an autosomal recessive inherited disorder, which means that two abnormal genes (one from each parent) are needed to have the disease. The defect causes the body to produce insufficient quantities of an enzyme called homogentisic acid oxidase. This enzyme normally breaks down a toxic byproduct of tyrosine metabolism called homogentisic acid. Although some of this is excreted in urine, small amounts remain in the body and slowly and progressively get deposited in bones and cartilage where it turns into a pigmented polymeric material.
Ochronosis may also be caused by external agents. These may include:
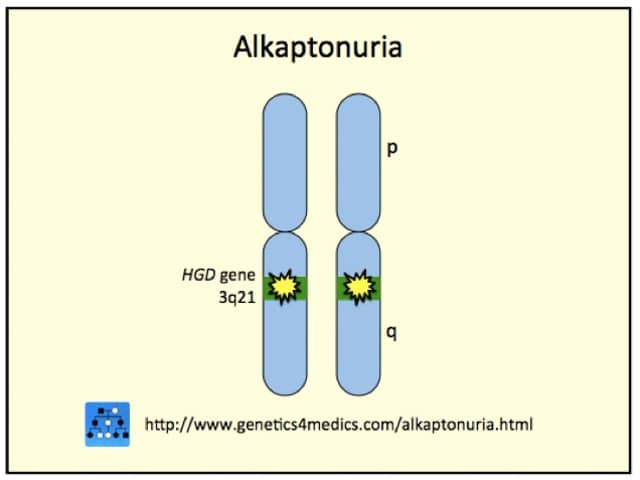
*Image courtesy Genetics 4 Medics
Most patients don't have any symptoms throughout childhood or early adult life and it is not until they reach their 40's that other signs of the disease start appearing. One of the earliest signs is thickening of the ear cartilage (the pinna feels noticeably thickened and flexible). In addition the skin turns a blue-black colour. Earwax is often reddish-brown or jet-black. Gradually patients will suffer sore joints, leading to arthropathy (joint disease characterised by swelling and enlarged bones). Many body parts become affected due to the build-up of pigment deposits in bones and cartilage.

Alkaptonuria is a lifelong disease. There is no cure for the condition. If alkaptonuria is diagnosed early on in life it is reasonable for patients to have a low-protein diet. This reduces the intake of amino acids phenylalanine and tyrosine, which in turn reduces the amount of homogentisic acid produced. Although not proven, this could potentially avoid or minimise complications later in life.
Vitamin C has been found to slow down the conversion of homogentisic acid to the polymeric deposits in cartilage and bone. A dose of up to 1g/day is recommended for older children and adults. Nitisinone, an enzyme inhibitor that mediates the formation of homogentisic acid is being used in restricted experimental treatments.
Life expectancy is normal although patients may be at increased risk of heart conditions and may require surgical treatments for spine, hip, knee and shoulder joint problems.
Exogenous cutaneous ochronosis has been successfully treated by laser.

