

Crouzon syndrome — extra information
Introduction Demographics Causes Clinical features Diagnosis Treatment Outlook
What is Crouzon syndrome?
Crouzon syndrome is characterised by a variety of craniofacial and developmental symptoms.
It is a hereditary condition inherited in an autosomal dominant pattern (an abnormal gene from one parent can cause the syndrome). It is also known as Crouzon disease, craniofacial dysostosis, craniostenosis, Apert–Crouzon syndrome, acrocephalosyndactyly type II, Vogt cephalosyndactyly, and trigorhinophalangeal dysplasia.
It was first described by Crouzon in 1912.
Who gets Crouzon syndrome?
Crouzon syndrome is rare, although it is still the most common craniosynostosis syndrome (where the fibrous joints of the skull prematurely close during infancy).
- It affects 1 in every 60,000 live births.
- It appears to be diagnosed equally in people of all races and ethnicities.
- It is often diagnosed at birth or in infancy due to distinctive facial features.
What causes Crouzon syndrome?
Crouzon syndrome is usually caused by mutations in the fibroblast growth factor receptor 2 (FGFR2) gene. The FGFR3 gene can also be involved.
- This mutation leads to signals to immature cells to become bone cells during embryogenesis.
- A family history of Crouzon syndrome is present in 50% of cases.
- In the other 50% of cases, the syndrome is sporadic, resulting from new gene mutations.
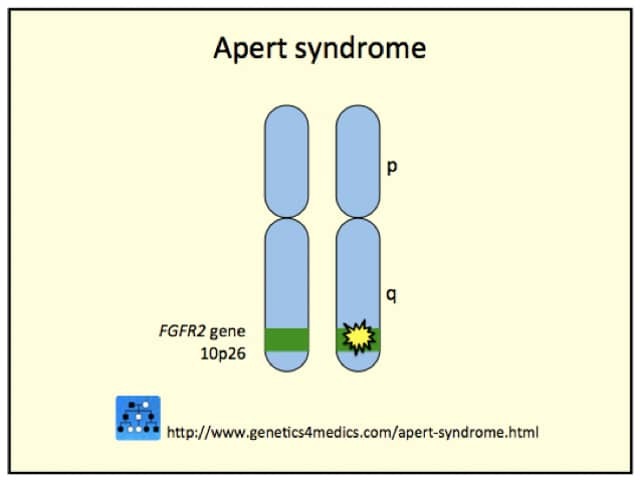
*Image courtesy Genetics 4 Medics
What are the clinical features of Crouzon syndrome?
The clinical features of Crouzon syndrome vary widely, and range from mild to severe. The key feature is the premature closure of the skullcap and cranial base sutures, and craniosynostosis.
Facial features
Related distinctive facial features include:
- Exophthalmos (abnormal protrusion of the eyeball)
- Hypertelorism (excessive width between the eyes)
- Hypoplastic maxilla (an underdeveloped jaw)
- Mandibular prognathism (protrusion of the lower jaw)
- A short upper lip
- A beaked nose.
Visual defects
Visual defects associated with Crouzon syndrome include:
- Amblyopia (dimness of sight without apparent change in the eye structures)
- Ametropia (refractory error)
- Strabismus (inability to attain binocular vision because of muscle imbalances in the eyeballs).
These visual defects are due to corneal injury, cataracts (a clouding of the lens of the eye or its surrounding transparent membrane), optic atrophy (deterioration of the optic nerve) and iris coloboma (a hole in the iris).
Other features
Other features associated with Crouzon syndrome include:
- Decreased mental function and risk of increased intracranial pressure and seizures
- Respiratory symptoms due to narrowing of the nasopharyngeal passage and deviated septum or other structural abnormalities
- Hearing loss and/or Ménière disease (a disorder of the inner ear characterised by episodes of vertigo, ringing in the ears, hearing loss, and pressure in the ear)
- Skeletal abnormalities, including the fusion of the spinal column.
The main dermatological sign of Crouzon syndrome is acanthosis nigricans, in which there is thickened, hyperpigmented skin with a velvety feel affecting the neck, torso, and face. It usually occurs by the onset of puberty.
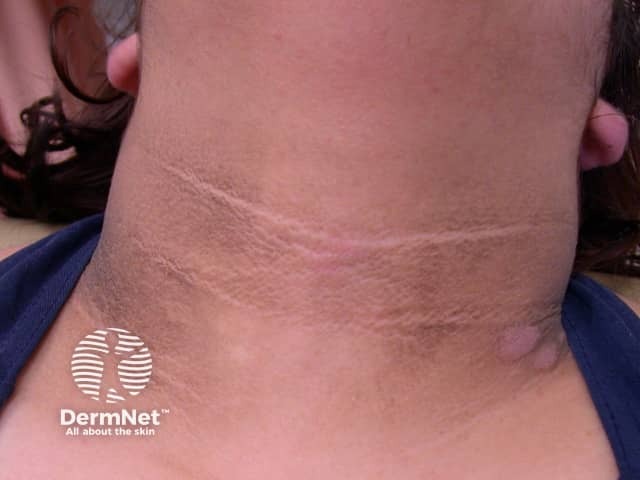
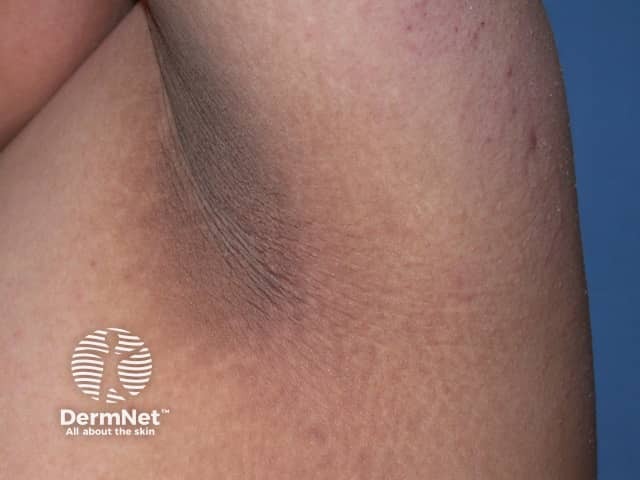
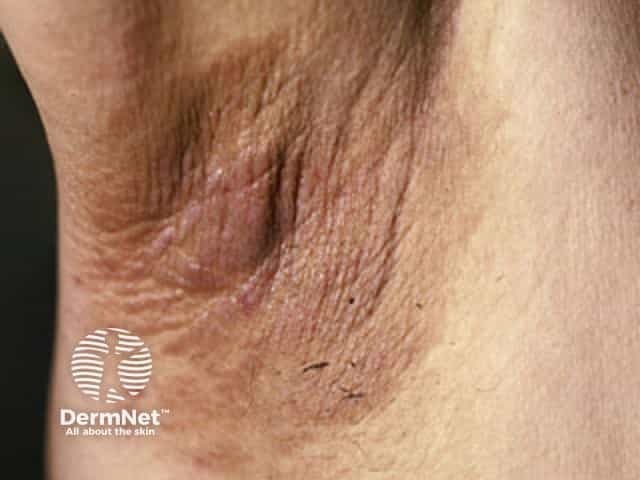
How is Crouzon syndrome diagnosed?
Crouzon is confidently diagnosed in a child with craniosynostosis when mutations on the FGFR2 gene are detected. The genes of the affected individual’s parents may also be tested to detect mosaicism (possessing cells of two or more genetically different types).
How is Crouzon syndrome treated?
The standard treatment for Crouzon syndrome includes early craniectomy (the surgical removal of a portion of the skull) and cosmetic reconstruction to help promote normal facial growth.
Multidisciplinary care may involve medical and surgical assessment and management of symptoms. This can include:
- Ophthalmological treatment of amblyopia, ametropia and strabismus
- Audiological treatment and myringotomy for hearing loss
- Shunting to treating intracranial pressure due to hydrocephalus (increased cerebrospinal fluid within the cranial cavity)
- Tracheostomy (an incision in the windpipe) to treat airway obstruction
- Orthodontic surgery to treat irregularities in the teeth and jaws.
What is the outlook for Crouzon syndrome?
Improvements in surgical techniques, specifically in craniofacial surgery, have greatly increased quality of life, intellectual and physical abilities, and social acceptance for children with Crouzon syndrome.
The lifespan for individuals with Crouzon syndrome is generally normal, but mortality can occur due to airway obstruction, acute respiratory distress, or increased intracranial pressure.
References
- Chen H; Medscape. Genetics of Crouzon syndrome. Updated 6 May 2015. Available at: http://emedicine.medscape.com/article/942989-overview (accessed July 2016).
- Genetics Home Reference, US National Library of Medicine. Crouzon syndrome. 2016. Available at: https://ghr.nlm.nih.gov/condition/crouzon-syndrome (accessed July 2016).
- National Organization for Rare Diseases (NORD).Crouzon syndrome. Updated 2015. Available at: http://rarediseases.org/rare-diseases/crouzon-syndrome (accessed July 2016).
On DermNet
Other websites
- Genetics of Crouzon syndrome — Medscape