Introduction Demographics Clinical features Diagnosis Treatment
Cryopyrin-associated periodic syndrome or syndromes (CAPS), also known as cryopyrinopathies, are genetic autoinflammatory syndromes defined by ‘gain-of-function’ mutations affecting the cryopyrin protein.
Three distinct clinical syndromes are recognised within CAPS.
The mildest form is FCAS, and the most severe form, NOMID/CINCA, is often fatal. Clinical overlap is now recognised, suggesting these are a continuum of disease.
Some cases of Schnitzler syndrome may also fall into this category.
Cryopyrin-associated periodic syndromes are all autosomal dominant genetic conditions, meaning only one copy of the abnormal gene is required to develop the clinical syndrome. FCAS and Muckle-Wells syndrome are usually familial, inherited from one affected parent. NOMID/CINCA appears as a spontaneous mutation, as it causes severe disabilities and is often fatal before adult life.
Patients with overlap features are increasingly reported:
Examples include cold-sensitivity, frequent headaches or asymptomatic papilloedema (swelling at the back of the eye) in MWD or deafness in FCAS. Even within the one family, members may be classified as having different syndromes based on clinical features.
All three conditions are caused by mutations in the same gene, NLRP3, located on chromosome 1 (1q44). Approximately 100 different mutations, mostly missense mutations in exon 3 of this gene, have been identified in patients with cryopyrin-associated periodic syndromes. Rarely missense mutations have been identified in exons 4 and 6. Certain mutations result in a specific clinical syndrome eg Y570C, Y570F, F309S, F523L are only reported in severe NOMID/CINCA. Other mutations (eg R260W, V198M, V262G, D303N), have been identified in more than one clinical picture, even within the one family. This would suggest there may be additional influences, either genetic or environmental, that affect the clinical presentation. Asymptomatic carriers, mostly in affected families, have also been detected.
The NLRP3 gene is expressed particularly in white blood cells (especially neutrophils) and chondrocytes (cartilage cells). It codes for cryopyrin, a protein involved in the formation of inflammasomes. Inflammasomes are protein complexes found inside cells. They are important in the innate immune system. Disease-linked changes in cryopyrin result in the loss of a regulatory step allowing increased caspase-1 activation and hence increased activation of interleukin (IL)-1. Activated IL-1β is a powerful stimulator of the inflammatory cascade. Expression of the NLRP3 gene in chondrocytes may be relevant for the joint pain/swelling in FCAS and MWS and the bony overgrowth in NOMID/CINCA. The latter does not appear to respond to IL-1 receptor blockade. Expression in chondrocytes may also be involved in the development of sensorineural deafness in MWS and NOMID/CINCA.
The mechanism by which cold triggers attacks in FCAS remains unknown.
Not all cases of clinically typical CAPS have had an NLRP3 gene mutation identified, particularly the NOMID/CINCA form. Somatic mutations, gene mutations occurring during fetal development, have been found to explain a small number of apparently mutation-negative cases. The NLRP3 mutation is then detected in only a percentage of cells in the peripheral blood. Such testing is difficult and currently done only in a research environment.
The clinical features common to these three conditions are intermittent or recurrent attacks comprising:
In addition, each syndrome has its own distinctive clinical features (see the description of a specific syndrome).
The urticaria-like rash is the same clinically and histologically in all forms of the cryopyrin-associated periodic syndromes. Characteristically:
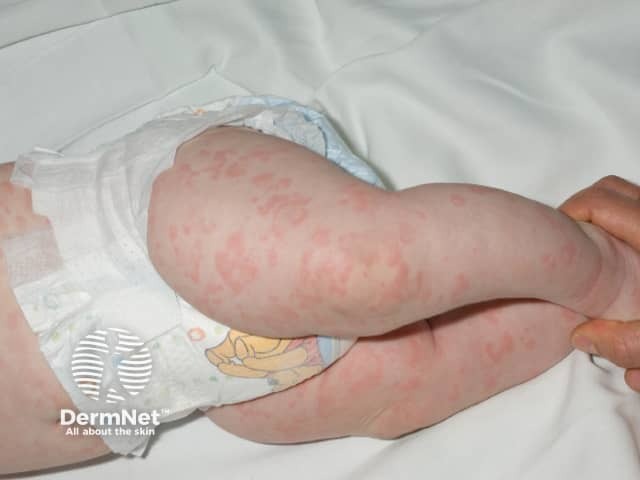
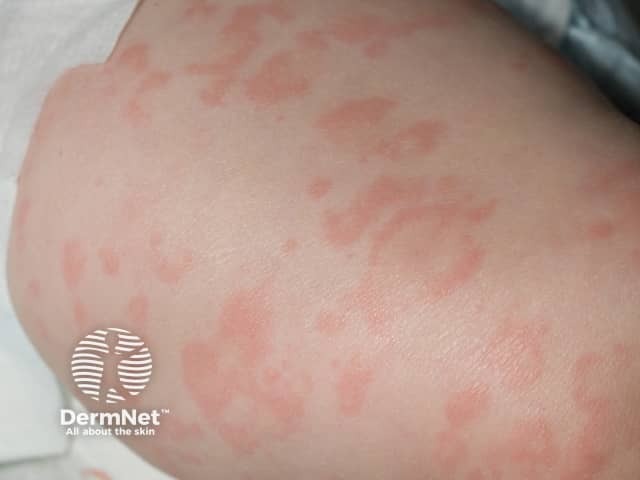
The diagnosis of a cryopyrin-associated periodic syndrome should be considered in patients presenting with recurrent episodes of fever, skin rash, joint pain and inflammation of the eyes, without evidence of infection or autoimmune disease. Although these are all autosomal dominant conditions, a positive family history may not always be elicited.
A delay in the diagnosis of a cryopyrin-associated periodic syndrome is common as all forms are rare. Initial diagnoses of viral infections and allergies are usually considered. This delay in diagnosis, and therefore in treatment, can result in interleukin-1-induced organ damage such as deafness and amyloidosis.
Acute phase reactants in blood, such as erythrocyte sedimentation rate (ESR), C-reactive protein (CRP) and serum amyloid A (SAA), are markedly increased, even when the patient is well between episodes. Anaemia of chronic disease and increased numbers of white blood cells, especially neutrophils, may be detected.
Skin biopsy from the urticaria-like rash can suggest the diagnosis of a cryopyrin-associated periodic syndrome, as it shows a perivascular and sometimes peri-eccrine (around sweat gland) neutrophilic infiltrate of the reticular dermis, without mast cells or vasculitis. This is quite different from normal urticaria (hives).
A commercial test is available for sequencing exon 3 of the NLRP3 gene. This will detect most, but not all known mutations associated with cryopyrin-associated periodic syndromes.
The three clinical syndromes all respond very well to interleukin-1 antagonist therapy, such as anakinra, rilonacept and canakinumab. All CAPS patients respond, provided a sufficient dose is administered.
The dramatic efficacy of the biologic agent anakinra, an IL-1 receptor antagonist, has been demonstrated in a number of large clinical trials involving many patients with cryopyrin-associated periodic syndromes. It is given as a daily subcutaneous injection, and local injection reactions (itching, swelling, redness) are the most common adverse effects. Anakinra is the standard of care for NOMID/CINCA and was approved by the FDA for this indication in 2013. The dose range varies: 0.5–1.5mg/kg/d for FCAS, up to 3.5 mg/kg/d for MWS and up to 10 mg/kg/d for severe NOMID/CINCA in infants.
Note: anakinra is not registered or subsidised in New Zealand (March 2011). In other countries such as the USA and Europe, its registered indication is rheumatoid arthritis.
Rilonacept was the first FDA-approved therapy for CAPS. It was approved in 2008 for FCAS and MWS in adults and children from the age of 12 years. Rilonacept is a dimeric fusion protein that binds to the interleukin-1 receptor accessory protein and IL-1 type 1 receptor. It is administered weekly as a subcutaneous injection. Adverse events have been reported in clinical trials as mild to moderate in severity, including local injection site reactions, upper respiratory tract infections, headache, joint pain and diarrhoea. Two deaths have been reported during rilonacept treatment for CAPS: one from pneumococcal meningitis and the other from coronary artery disease.
Canakinumab has also been FDA-approved (2009) for FCAS and MWS and has been approved from the age of 4 years. In Europe, it has been approved for all forms of CAPS. Canakinumab is a fully humanised monoclonal antibody specifically targeted against IL-1β. It is administered as a subcutaneous injection every 8 weeks. In a clinical trial involving 35 CAPS patients, the commonly reported side effects were nasopharyngitis, rhinitis, nausea, diarrhoea and vertigo. Local injection site reactions were not reported. Two serious adverse events reported during trials were vertigo with acute angle glaucoma and recurrent lower urinary tract infection with sepsis. Vertigo may develop more commonly in patients with the sensorineural deafness associated with some forms of CAPS.
A dramatic response is seen within hours to days of the injection for the daily symptoms (fevers, rash, headaches, joint pain, conjunctivitis) and acute phase reactants in blood. Improvement of long-term complications have also been reported in some patients with hearing loss, loss of vision, amyloidosis (kidney disease), growth retardation, but not the bone abnormalities or existing intellectual disability seen in severe NOMID/CINCA. There is evidence to suggest that starting treatment in very young children may prevent or minimise long-term neurologic disability.
It has been recommended that the long-acting anti-IL-1 agents in very young children should be used in conjunction with antibiotic prophylaxis and immunisation against Streptococcus pneumoniae and Haemophilus influenzae as for post-splenectomy.
Treatments that block the effect of IL-1beta have had a dramatic effect on the quality of life for sufferers of these rare cryopyrin-associated periodic syndromes.

