Bullous pemphigoid is the most common autoimmune dermatosis presenting with crops of tense pruritic blisters, often in older adults. Mucosal involvement may occur and a number of clinical subtypes exist. Autoantibodies are directed to components of the basement membrane, particularly the BP antigens BP180 and BP230.
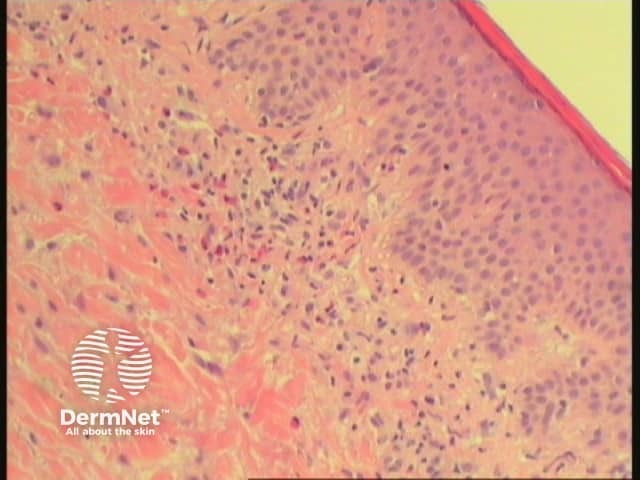
Early lesions during the urticarial phase may be non-specific and show mild upper dermal oedema with perivascular infiltrate (figure 1). There are often conspicuous eosinophils. Immunofluorescence is very helpful in this early phase.
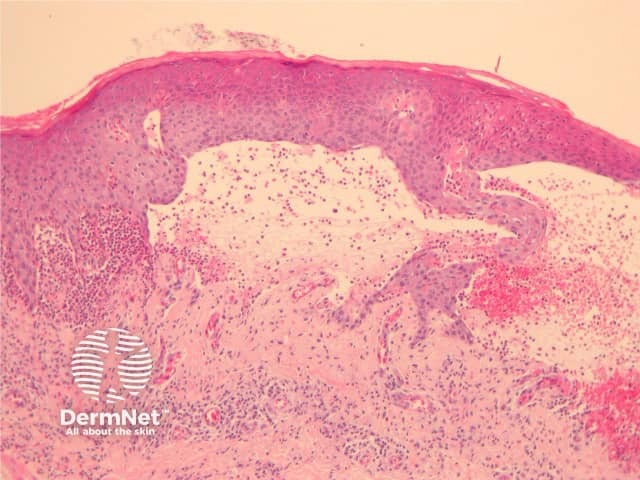
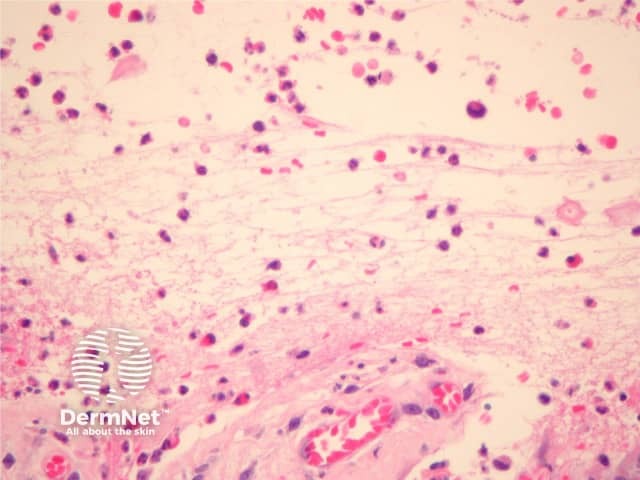
Established blister: The blister is subepidermal and contains fibrin and large numbers of inflammatory cells including eosinophils (figures 2, 3). The eosinophils may be very dense in areas and form small abscesses in the superficial dermis.
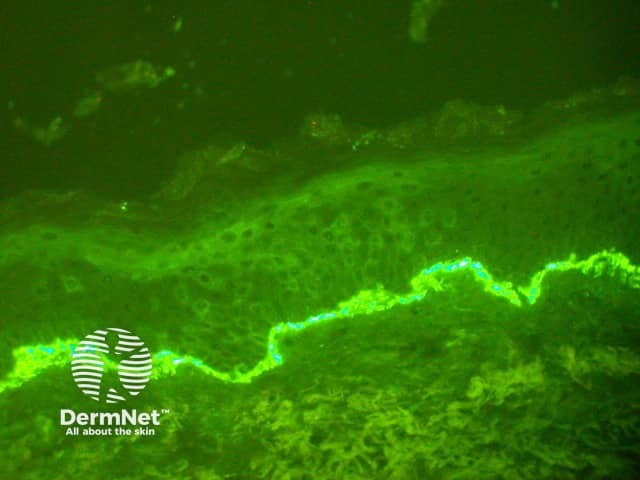
Direct immunofluorescence shows linear deposition of IgG (most often IgG4 subtype) and C3 along the basement membrane (figure 4). Split skin indirect immunofluorescence studies are generally not needed, but may be useful in rare cases when epidermolysis bullosa aquisita is suspected.
Dermatitis herpetiformis: Neutrophil dermal papillary microabscesses seen in dermatitis herpetiformis are not usually present in bullous pemphigoid but may be present in some variants. Direct immunofluorescence shows granular deposition of IgA.
Linear IgA disease: Subepidermal blister formation with linear basement membrane IgA deposition on direct immunofluorescence.
Epidermolysis bullosa aquisita shows subepidermal bullae with linear IgG/C3 deposition however indirect immunofluorescence in these condition reveals positivity on the floor of split skin whereas bullous pemphigoid has positivity on the epidermal side.
Other causes of subepidermal blistering include epidermolysis bullosa, porphyria cutanea tarda and bullous amyloidosis.

