Introduction Demographics Diagnosis Treatment Specific types Other types
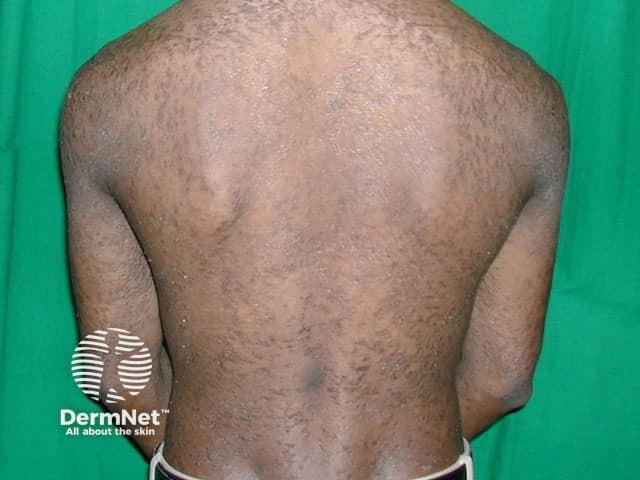
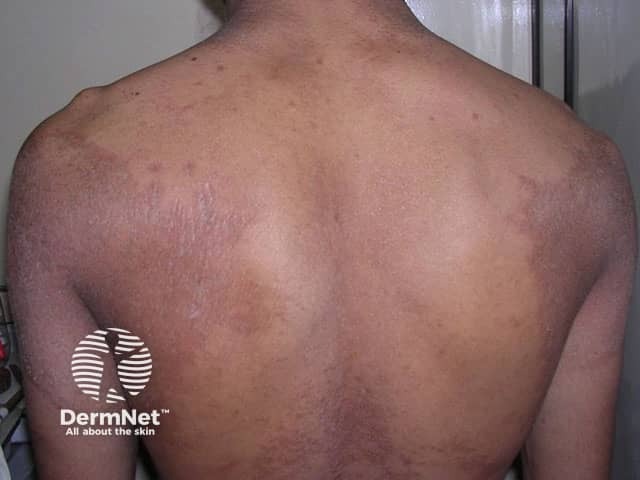
Erythrokeratoderma, sometimes called erythrokeratodermia, is the descriptive name given to a rare group of disorders of keratinisation. This is the process that forms the different layers of the epidermis, the outermost layer of the skin. The various erythrokeratodermas are characterised by well-demarcated plaques of erythema (redness) and hyperkeratosis (scaling).
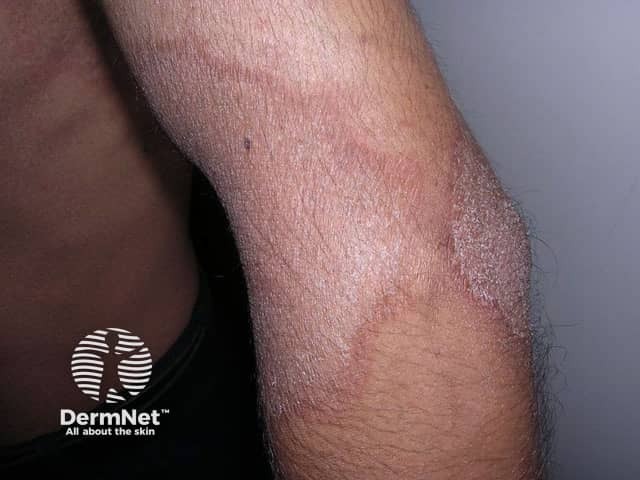
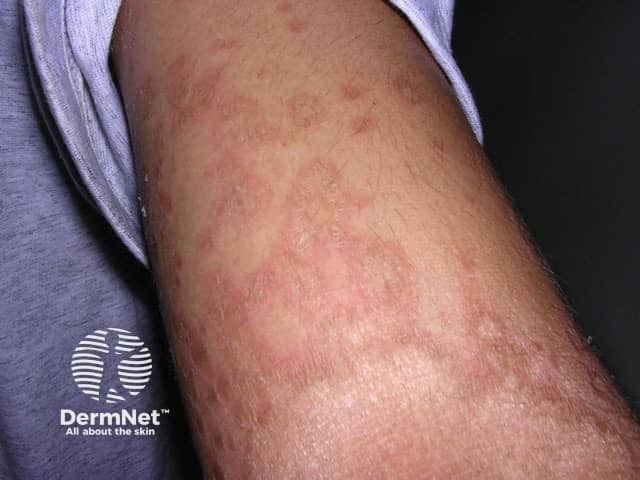
Erythrokeratoderma is due to several genetically inherited disorders, mostly autosomal dominant. This means that the gene comes from one parent and that an individual with the disease may pass it on to 50% of his or her children. Sporadic cases occur due to new genetic mutations at conception.
In most types of erythrokeratoderma, the underlying defect appears to be a mutation in one of the connexin genes. Connexins are gap junction proteins, found in the channels that connect adjacent cells. Different connexins are found in different tissues, accounting for variability in presentation.
Erythrokeratoderma is diagnosed by its clinical appearance. A skin biopsy can be performed for histology, but there are no distinctive features.
Genetic counselling should be offered to affected individuals and their families of childbearing age. In time, genetic tests for the specific disorders may be available to some families.
There is no specific or curative treatment for erythrokeratoderma. Minimising temperature changes and mechanical friction is important. Symptomatic improvement can be obtained by:
Because erythrokeratoderma is rare, the classification of the different types is still evolving. There are a few well-defined syndromes and some other atypical variants which are listed below.
Erythrokeratoderma variabilis is the commonest of the erythrokeratodermas and is autosomal dominantly inherited or sporadic. More than 50% of the affected individuals show skin lesions at birth or during the neonatal period and 90% will show some evidence of the disease within their first year of life.
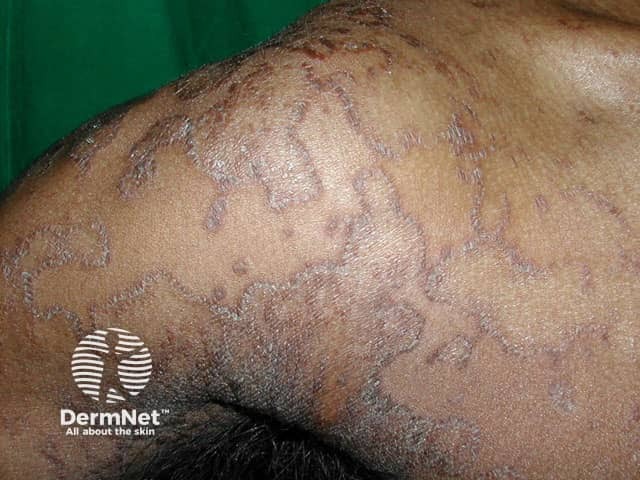
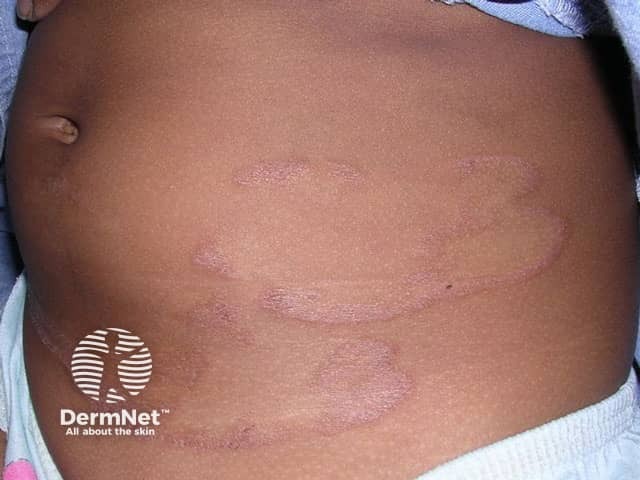
Erythrokeratoderma variabilis results in well-defined round or oval red, scaly plaques (thick patches) that may join together to form map-like patterns. There are 2 types of skin lesion:
In severe cases, the erythrokeratoderma can be generalised.
Some patients with erythrokeratoderma variabilis experience burning sensations and itching in the affected areas and others have no symptoms. The skin lesions may be triggered by internal and/or external factors, such as:
The hair, teeth and nails are not involved. Erythrokeratoderma does not affect physical and mental development and the general health of the individual is not affected.
After gradual progression throughout infancy and childhood, the disease tends to stabilise during puberty.
Erythrokeratoderma progressiva symmetrica is a very rare form of erythrokeratoderma that is also genetically inherited. Sporadic cases may occur.
Skin lesions are not present at birth and begin during infancy or early childhood. Affected individuals show fixed or slowly progressive red, scaly plaques symmetrically distributed on the body. The extremities, i.e., hands and feet, are often involved, which is uncommon in erythrokeratoderma variabilis.
Physical and mental development are normal.
Like erythrokeratoderma variabilis, the skin lesions of erythrokeratoderma progressiva symmetrica slowly progress and increase in number and size during childhood, and tend to stabilise after puberty. The inherited cases have symptoms lifelong but there are reports of spontaneous improvement after many years in the sporadic cases.
Progressive partially symmetrical erythrokeratodermaresults in deafness, muscle weakness, peripheral nerve damage, physical and intellectual disability in addition to atypical peripheral erythrokeratoderma.
Other types of erythrokeratoderma are very rare and only a few cases have been reported worldwide. Many of these had only skin lesions and were otherwise well.
Progressive partially symmetrical erythrokeratoderma results in deafness, muscle weakness, peripheral nerve damage, physical and intellectual disability in addition to atypical peripheral erythrokeratoderma.

