Introduction
Causes
Clinical features
Complications
Management
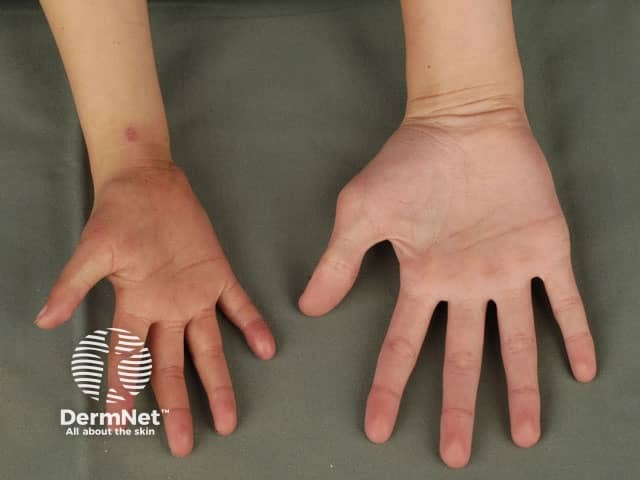
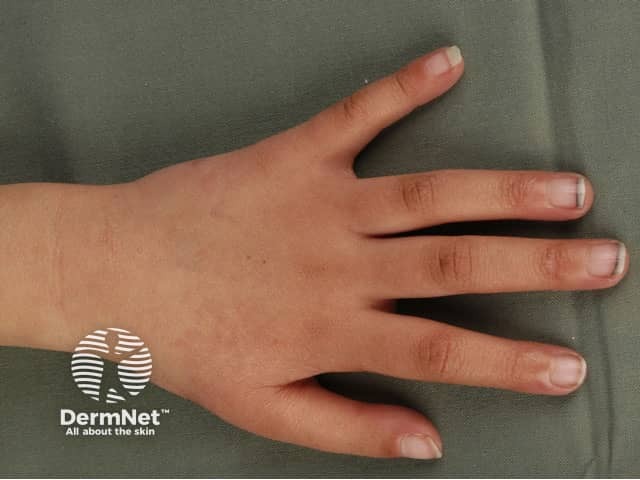
Rothmund–Thomson syndrome is a rare inherited disease that affects the skin, eyes, bones and internal organs. At least 300 cases have been reported in medical journals since a case was first described by Rothmund in 1868. Rothmund–Thomson syndrome is also known as poikiloderma congenitale.
Rothmund–Thomson syndrome is due to a genetic defect, in which there are mutations in the RECQL4 gene on Chromosome 8. This gene encodes for the enzyme DNA helicase which unwinds DNA (deoxyribonucleic acid). The abnormal gene makes the chromosomes unstable, altering the growth of cells in many tissues.
The defect is inherited as an autosomal recessive trait. This means an abnormal gene must come from each parent.
Affected children may be identified early in life by their small size, their tendency to sunburn easily, and from the appearance of their skin, teeth and bones. Rothmund–Thomson syndrome is slightly more common in females than males.
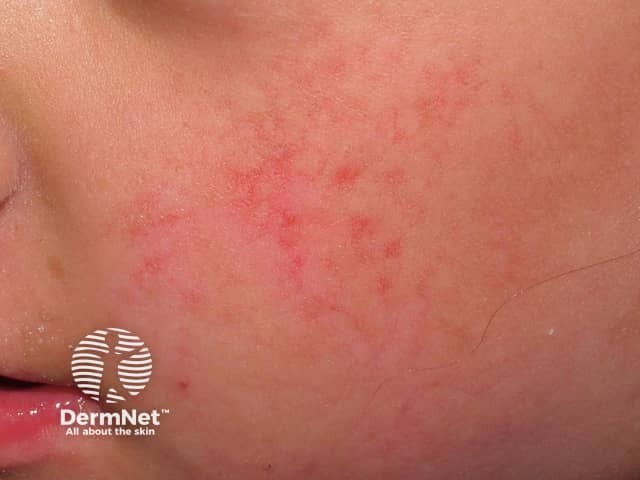
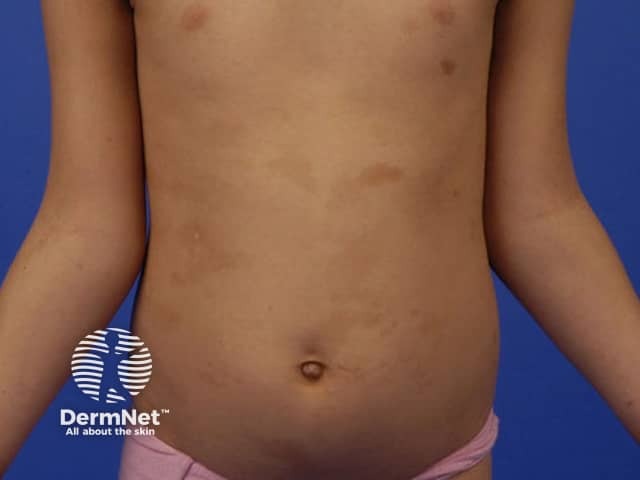
Skin
Eyes
Bones
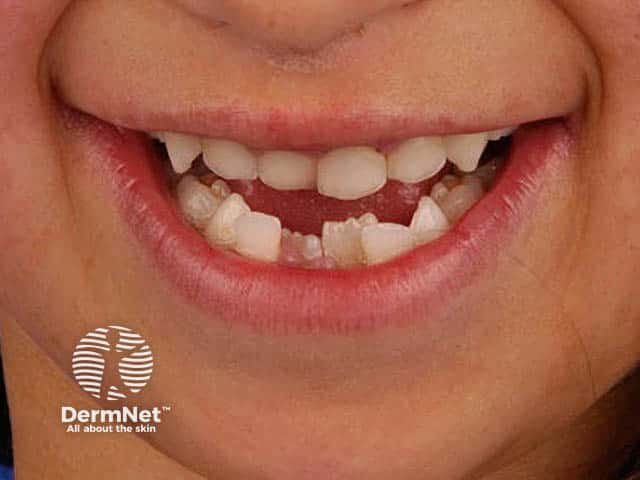
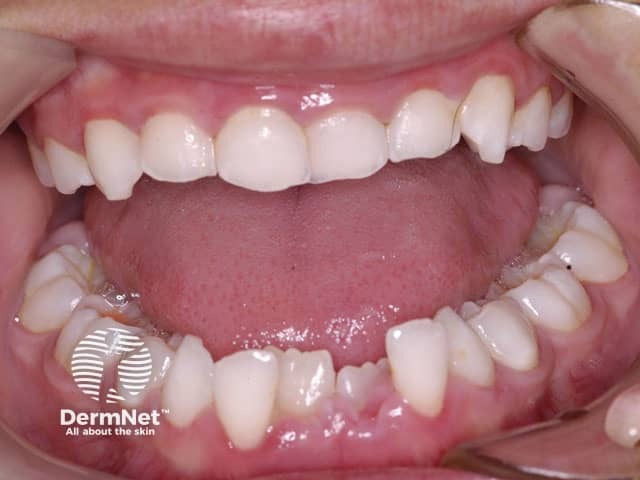
Dentition
Reproductive system
Endocrine system
Gastrointestinal system
Intellect
The main concern in Rothmund–Thomson syndrome is an increased susceptibility to cancer.
Skin cancers including basal cell carcinoma, squamous cell carcinoma and intraepidermal carcinoma (Bowen disease) are common in older children and adults with Rothmund–Thomson syndrome. They often arise on the face, neck and limbs. It is postulated that skin cancers occur because of defects in DNA repair after exposure to ultraviolet radiation.
The second most common type of cancer is osteosarcoma, which may develop in late childhood or adolescence. Osteosarcoma may arise within pre-existing bone dysplasia so it may be difficult to diagnose on X-ray.
Treatment of osteosarcoma in Rothmund–Thomson syndrome is similar to in patients without the syndrome.
Other cancers affecting individuals with Rothmund–Thomson syndrome include:
Children with Rothmund–Thomson syndrome are often followed up by a paediatrician, dermatologist, orthopaedic surgeon, dental surgeon and/or other specialists. Clinicians should bear in mind the risk of cancers and should monitor and investigate as appropriate.
Sun protection is very important because of photosensitivity and increased risk of skin cancer. This should include seeking shade, fully covering clothing and broad-spectrum sunscreens.
Genetic counselling is important for family members.

