Cobimetinib is a prescription medicine used to treat melanoma that has spread to other parts of the body, cannot be removed by surgery, and has an abnormal BRAF gene. It should not be used to treat melanoma in patients with a normal or wild-type BRAF gene.
In 2015, the US Food and Drug Administration (FDA) approved cobimetinib (Cotellic™, Genentech Inc. California, USA) in combination with vemurafenib (a specific BRAF–kinase inhibitor) for the treatment of melanoma.
In the same year, the Committee for Medicinal Products for Human Use at the European Medicines Agency issued a positive opinion for marketing authorization for cobimetinib in the European Union.
Cobimetinib has since received approval for marketing in New Zealand through Medsafe in 2017 for the treatment of patients with melanoma.
FDA approval was based on results from the Phase III CoBRIM study, which showed cobimetinib, when used in combination with vemurafenib, reduced the risk of disease worsening or death by about half in patients with melanoma.
CoBRIM was an international, randomised, double-blind, placebo-controlled Phase III study that evaluated the safety and efficacy of 60 mg of cobimetinib once daily plus 960 mg twice daily of vemurafenib compared to 960 mg of vemurafenib twice daily plus placebo. A total of 495 patients were involved in the study, all of whom had previously untreated BRAF V600 mutation-positive, unresectable, locally advanced or metastatic melanoma.
The presence of BRAF V600 mutation was detected using the cobas®; 407 4800 BRAF V600 mutation test.
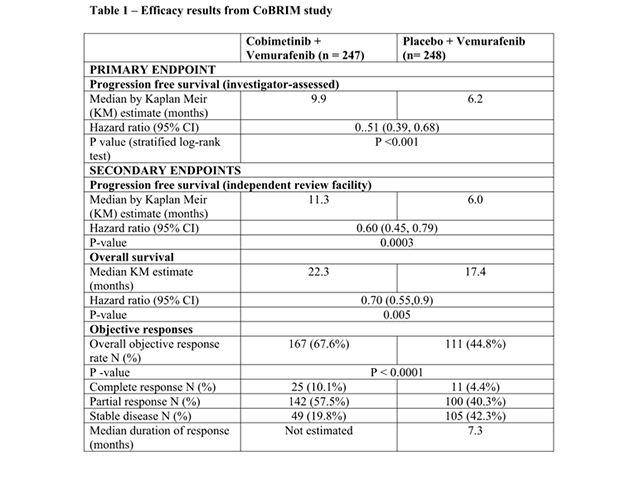
Efficacy results are summarised in Table 1.
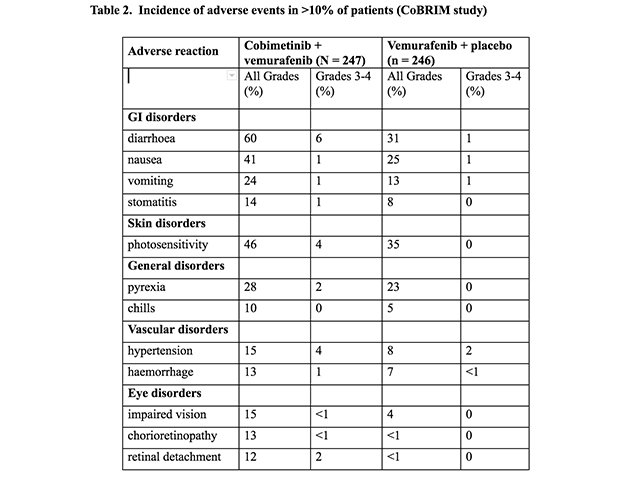
The most common (≥ 20%) adverse reactions with cobimetinib were:
Because clinical trials are conducted under widely varying conditions, the adverse reaction rates observed in clinical trials may not reflect the rates observed in practice. In the CoBRIM study, 15% of patients receiving cobimetinib experienced an adverse reaction that resulted in permanent drug discontinuation.
The most common adverse reactions resulting in permanent drug discontinuation were:
Among the 247 patients receiving cobimetinib, adverse reactions led to dose interruption or reductions in 55%. The most common reasons for dose interruptions or reductions of cobimetinib were:
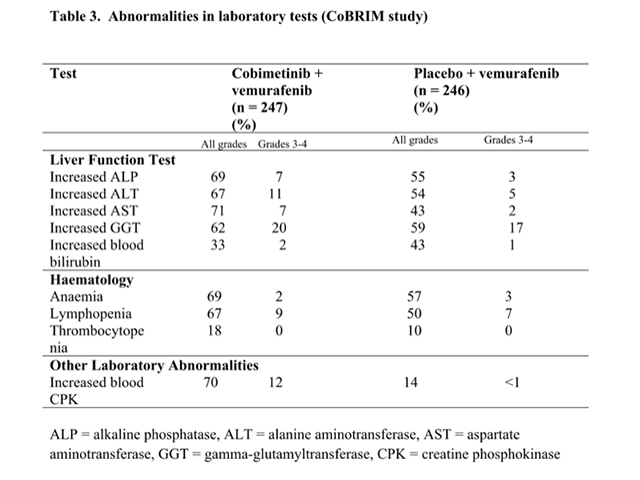
Reporting suspected adverse reactions after the authorisation of a medicine is important, as it allows continued monitoring of the benefit/risk balance of the medicine. Healthcare professionals in New Zealand are advised to report any suspected adverse reactions to the New Zealand Pharmacovigilance Centre.
There is no experience with cobimetinib overdose in human clinical trials. In case of suspected overdose, cobimetinib should be withheld and supportive care instituted. There is no specific antidote for overdosage with cobimetinib.
The New Zealand National Poisons Centre should be contacted on 0800 764 766 for advice on the management of overdose.
Approved datasheets are the official source of information for medicines, including approved uses, doses, and safety information. Check the individual datasheet in your country for information about medicines.
We suggest you refer to your national drug approval agency such as the Australian Therapeutic Goods Administration (TGA), US Food and Drug Administration (FDA), UK Medicines and Healthcare products regulatory agency (MHRA) / emc, and NZ Medsafe, or a national or state-approved formulary eg, the New Zealand Formulary (NZF) and New Zealand Formulary for Children (NZFC) and the British National Formulary (BNF) and British National Formulary for Children (BNFC).

