Introduction Demographics How it works Administration Use in specific populations Side effects Contraindications
Cobimetinib (Cotellic™, Genentech Inc. California, USA) is a prescription medication used, in combination with vemurafenib, for the treatment of patients with melanoma.
Cobimetinib received US Food and Drug Administration (FDA) approval in the US in 2015. In April 2017, Medsafe approved cobimetinib for the treatment of melanoma patients in New Zealand.
Cobimetinib, in combination with vemurafenib, is indicated for the treatment of patients with unresectable or metastatic melanoma, in which the cancer that has spread to other parts of the body or cannot be removed by surgery. An indicator of this melanoma is an abnormal gene called the BRAF V600E or V600K mutation, which must be confirmed in tumour specimens prior to initiation of treatment.
Cobimetinib is not indicated for treatment of patients with a normal or wild-type BRAF gene.
Cobimetinib is a small molecule inhibitor that blocks the MEK enzyme, a component of the kinase cascade in the mitogen–activated protein kinase (MAPK) pathway. Components of the MAPK pathways are frequently mutated in patients with malignant melanoma, particularly the RAF isoform BRAF. Mutations of BRAF cause a constitutive activation of these signalling pathways, which can lead to cancer.
Cobimetinib and vemurafenib target two different kinases in the MAPK pathway. Clinical trials have shown that the combination of cobimetinib with vemurafenib results in improvements in survival in patients with melanoma who harbour the BRAF V600 mutation.
Cobimetinib is available in 20-mg tablets under the brand name Cotellic.
Vemurafenib (ZELBORAF™) should be taken every 12 hours for every day in the 28-day cycle (no rest period). If a dose of vemurafenib is missed, it should be taken as soon as remembered. Do not make up for the missed dose if the missed dose is within 4 hours of the next scheduled dose.
No data are available for the use of cobimetinib in pregnant women to inform of a drug-associated risk for major birth defects and miscarriage. Based on findings from animal reproduction studies and its mechanism of action, cobimetinib can cause fetal harm. Pregnant women should be advised of the potential risk to a fetus.
There is no information on the presence of cobimetinib in human milk or its effects on a breastfed infant. The risk–benefit potential should be considered when prescribing cobimetinib to the mother. Nursing women should be advised not to breastfeed during treatment with cobimetinib and for 2 weeks after the final dose.
The safety and effectiveness of cobimetinib have not been evaluated in children.
Female patients of reproductive potential should be advised to use effective contraception during treatment with cobimetinib and for 2 weeks after the final dose. Based on findings in animals, cobimetinib may reduce fertility in females and males of reproductive potential.
Clinical studies with cobimetinib did not include sufficient numbers of individuals aged 65 years and over to determine whether they respond differently from younger subjects.
Dose adjustment of cobimetinib is not required in patients with mild (Child–Pugh score A), moderate (Child–Pugh B) or severe (Child–Pugh C) hepatic impairment. No dedicated pharmacokinetic trial in patients with renal impairment has been conducted. Dose adjustment is not recommended for mild–to–moderate renal impairment (creatinine clearance 30–89 mL/minute) based on the results of the population pharmacokinetic analysis. A recommended dose has not been established for patients with severe renal impairment.
Common side–effects include:
Uncommon, but potentially severe side–effects include:
Elevations in serum aminotransferase and alkaline phosphatase levels are common during vemurafenib therapy, and are even more common when it is combined with cobimetinib.
Instances of clinically apparent liver injury with jaundice have been reported during the clinical trials of cobimetinib and vemurafenib therapy, but the clinical features, course and outcomes of these episodes have not been described in detail.
The MAPK pathway inhibitors as a class are often associated with transient serum enzyme elevations and, more rarely, with instances of clinically apparent liver injury, but the clinical features have not been described and the association with cobimetinib has not been clearly defined.
The rate of clinically significant liver injury and hepatic failure associated with protein kinase inhibitors is increased in patients with pre–existing cirrhosis or hepatic impairment due to liver tumour burden. The product label for cobimetinib recommends monitoring of routine liver function tests during treatment.
Serum aminotransferase elevations above five times the upper limit of normal (if confirmed) should lead to temporary discontinuation, which should become permanent if these laboratory values do not improve significantly or resolve within a few weeks.
There does not appear to be cross–reactivity with other tyrosine kinase receptor inhibitors. In some situations, switching to another protein kinase inhibitor may be appropriate.
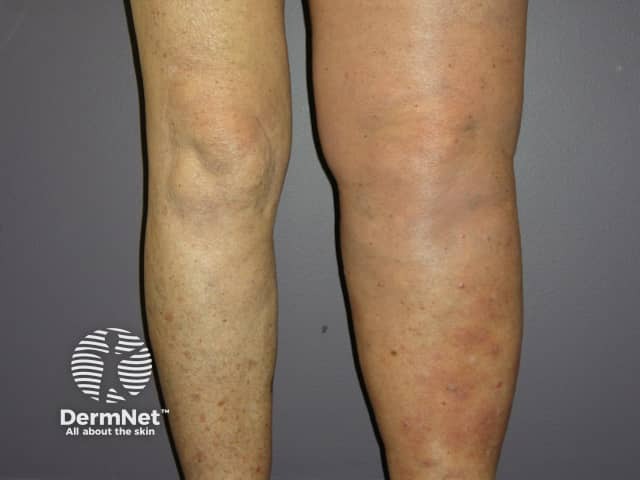
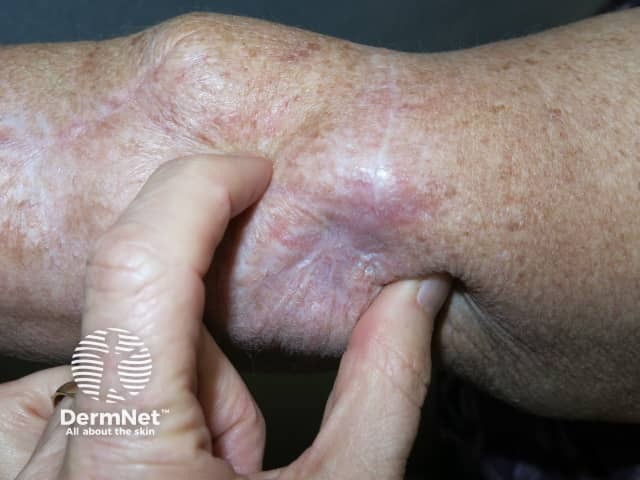
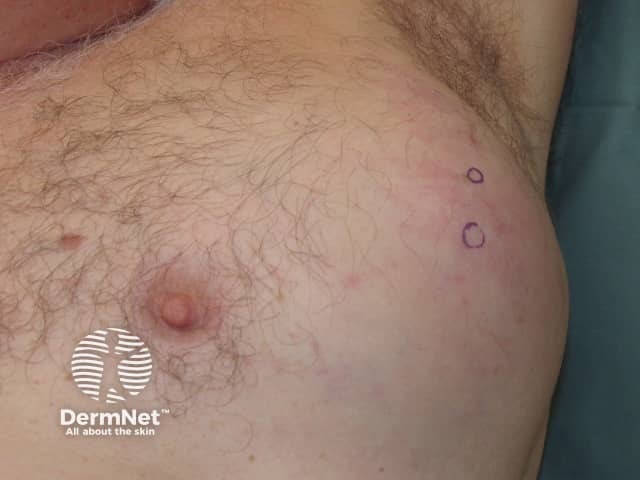
Cobimetinib in combination with vemurafenib may cause new skin cancers. These can include:
Cardiomyopathy, defined as symptomatic and asymptomatic decline in left ventricular ejection fraction (LVEF), can occur with cobimetinib.
The safety of cobimetinib has not been established in patients with a baseline LVEF that is either below the institutional lower limit of normal or below 50%.
LVEF should be evaluated prior to initiation of cobimetinib, 1 month after initiation, and every 3 months thereafter until drug discontinuation. Events of left ventricular dysfunction should be managed through treatment interruption, reduction, or discontinuation.
Cobimetinib is metabolised in the liver via the cytochrome P450 system, predominantly CYP3A, and is susceptible to drug–drug interactions with strong inhibitors or inducers of this microsomal enzyme.
Co–administration of cobimetinib with itraconazole (a strong CYP3A4 inhibitor) increased cobimetinib systemic exposure by 6.7-fold.
Concurrent use of cobimetinib and strong or moderate CYP3A inhibitors should be avoided. If concurrent short–term use (14 days or less) of moderate CYP3A inhibitors including certain antibiotics (eg, erythromycin, ciprofloxacin) is unavoidable (14 days or less), the dose of cobimetinib should be reduced from 60 to 20 mg.
An alternative to a strong or moderate CYP3A inhibitor should be used in patients who are taking a reduced dose of cobimetinib (40 or 20 mg daily). After discontinuation of a moderate CYP3A inhibitor, cobimetinib should be resumed at the previous dose.
Co–administration of cobimetinib with a strong CYP3A inducer may decrease cobimetinib systemic exposure by more than 80% and reduce its efficacy. Concurrent use of cobimetinib and strong or moderate CYP3A inducers including but not limited to carbamazepine, efavirenz, phenytoin, rifampicin, and St John's wort should be avoided.
Cobimetinib should be withheld in the following circumstances:
Approved datasheets are the official source of information for medicines, including approved uses, doses, and safety information. Check the individual datasheet in your country for information about medicines.
We suggest you refer to your national drug approval agency such as the Australian Therapeutic Goods Administration (TGA), US Food and Drug Administration (FDA), UK Medicines and Healthcare products regulatory agency (MHRA) / emc, and NZ Medsafe, or a national or state-approved formulary eg, the New Zealand Formulary (NZF) and New Zealand Formulary for Children (NZFC) and the British National Formulary (BNF) and British National Formulary for Children (BNFC).

