Introduction
How it works
Demographics
Detection of BRAF-600 mutation
Key clinical-trial evidence
Patients with BRAF V600E mutation-positive melanoma with brain metastases
Cobimetinib combined with vemurafenib in advanced BRAF (V600)-mutant melanoma (coBRIM)
Dosage and administration
Adverse events
Cutaneous adverse reactions
Warnings and precautions
Drug interactions
Use
Future considerations
Vemurafenib (ZELBORAF®) is a kinase inhibitor indicated for the treatment of patients with unresectable or metastatic melanoma with BRAF V600E mutation as detected by an FDA-approved test.
In August 2011, the US Food and Drug Administration (FDA) approved vemurafenib for the first-line treatment of both metastatic and unresectable (inoperable) melanoma. Vemurafenib was registered by MedSafe for use in New Zealand in February 2012 but is not currently funded by PHARMAC (August 2018). It can also be taken in combination with cobimetinib, a MEK inhibitor.
Vemurafenib and vemurafenib with cobimetinib have demonstrated prolonged survival in advanced melanoma patients.
Vemurafenib is a threonine kinase inhibitor, one of a new class of medicines known as epidermal growth factor receptor (EGRF) inhibitors or targeted therapy.
Melanoma tumours that carry the BRAF V600 mutation can be identified by a companion diagnostic test developed by the pharmaceutical company Roche, USA, and approved by the FDA.
The test is called the cobas® 4800 BRAF V600 Mutation Test and must be performed on melanoma biopsy samples from all patients before starting treatment with vemurafenib.
The test has several advantages including:
The FDA approval of vemurafenib was based on results from two clinical studies (BRIM3 and BRIM2) in patients with BRAF V600E mutation-positive, inoperable or metastatic melanoma as determined by the cobas® BRAF Mutation Test.
BRIM3 (B-RAF Inhibitor in Melanoma phase 3) was a global, randomised, open-label, multicenter, advanced (phase III) study that compared 960 mg of vemurafenib given orally twice daily with dacarbazine (previous standard of care for advanced melanoma treatment) 1000 mg/m2 given IV on day 1, every 3 weeks in 675 patients with previously untreated BRAF V600E mutation-positive, unresectable (inoperable) or metastatic melanoma. Treatment continued until disease progression, unacceptable toxicity, and/or consent withdrawal.
Key results are tabulated below:
Vemurafenib |
Dacarbazine |
|
|---|---|---|
Overall survival (OS) |
||
Number of Deaths |
78 (23%) |
121 (36%) |
Median OS (months) |
13.6 |
10.3 |
Median Follow-up (months) |
13.4 |
9.2 |
Progression-free survival (PFS) |
||
Median PFS (months) |
5.3 |
1.6 |
Complete tumour shrinkage (% patients) |
1% |
0 |
Partial tumour shrinkage (% patients) |
47.4% |
5.5% |
BRIM2 was a global, single-arm, multicentre, open-label early-phase (phase II) study that enrolled 132 patients with previously treated BRAF V600 mutation-positive metastatic melanoma. The primary endpoint of the study was the best overall response rate.
Data showed that:
Key results are tabulated below.
% Patients |
Cohort A (N = 90) |
Cohort B (N = 56) |
|
|---|---|---|---|
Confirmed best overall response rate in brain |
18% |
18% |
|
Complete response |
2% |
0% |
|
Partial response |
16% |
18% |
|
The median duration of response, months (95%CI; Kaplan Meier estimate) |
4.6 (2.9, 6.2) |
6.6 (2.8, 10.7) |
|
Vemurafenib has not been studied in patients with wild-type BRAF melanoma.
The presence of BRAF V600E mutation in tumour specimens should be confirmed prior to initiation of treatment with vemurafenib.
There are only limited data about the safety and efficacy of vemurafenib due to small patient numbers treated up to now. Treatment should be monitored including monthly liver function tests and as clinically indicated.
The most common adverse reactions (≥ 10% treated patients) reported with vemurafenib in BRIM3 and BRIM2 trials were:
Management of symptomatic adverse drug reactions may require dose reduction, treatment interruption, or treatment discontinuation. Dose reductions resulting in a dose below 480 mg twice daily are not recommended.
Some of the cutaneous side effects of vemurafenib are similar to those described with other EGFR and protein kinase inhibitors, but cutaneous squamous cell carcinoma (SCC) appears to be a specific problem. Concern has been expressed that vemurafenib may also lead to more melanocytic lesions, including common moles and perhaps new primary melanoma. The reactions are dose-related and may settle down even with continued therapy. Cutaneous side effects include:
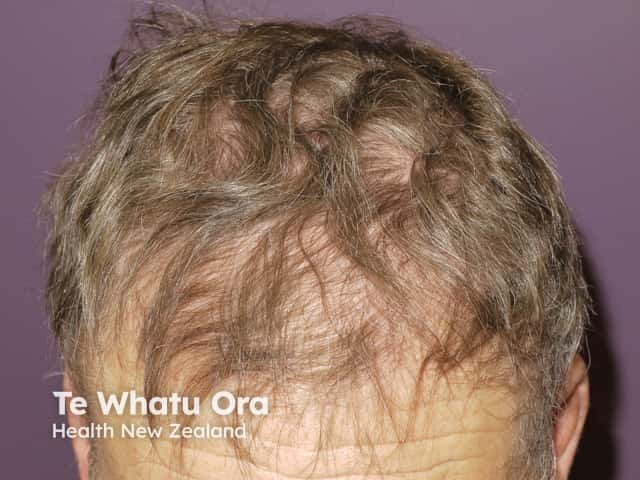

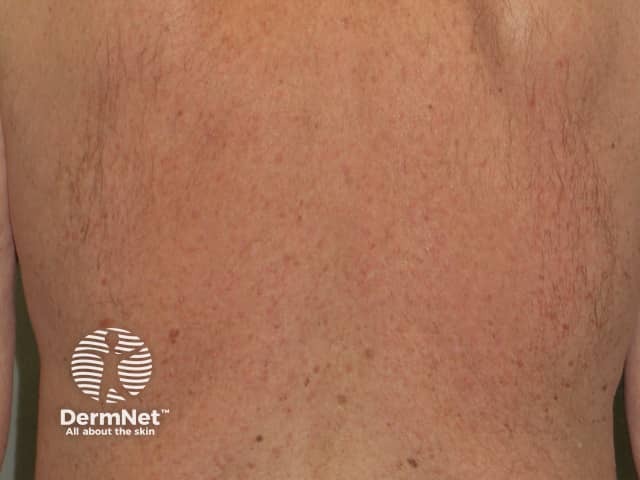
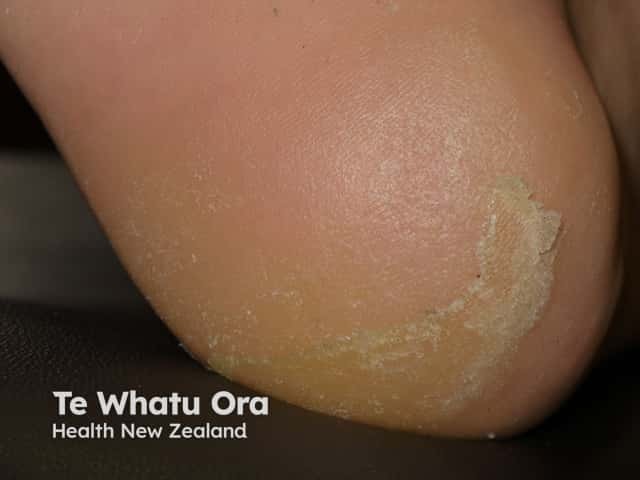
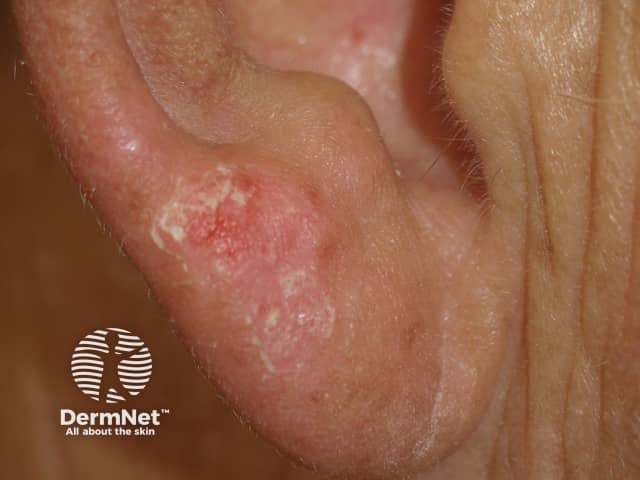
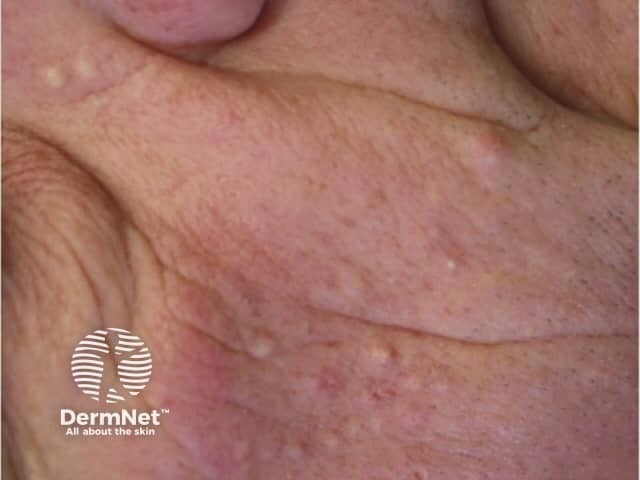
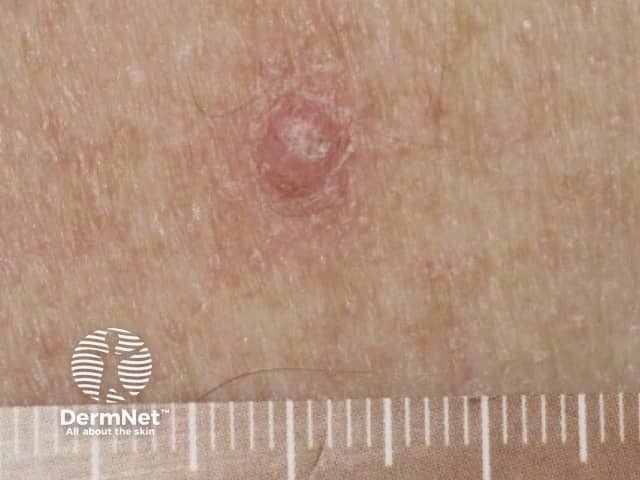
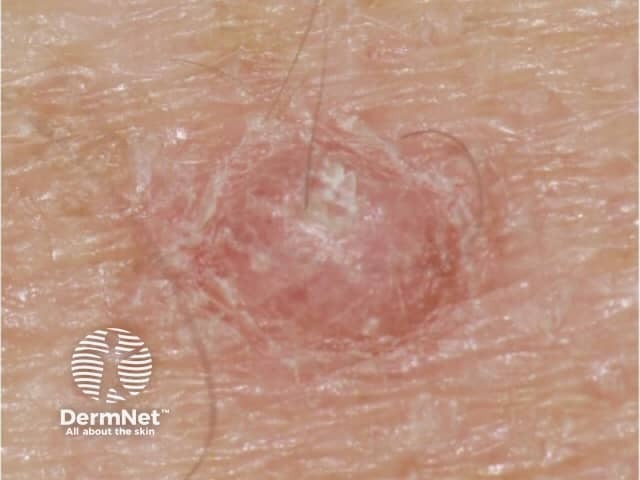
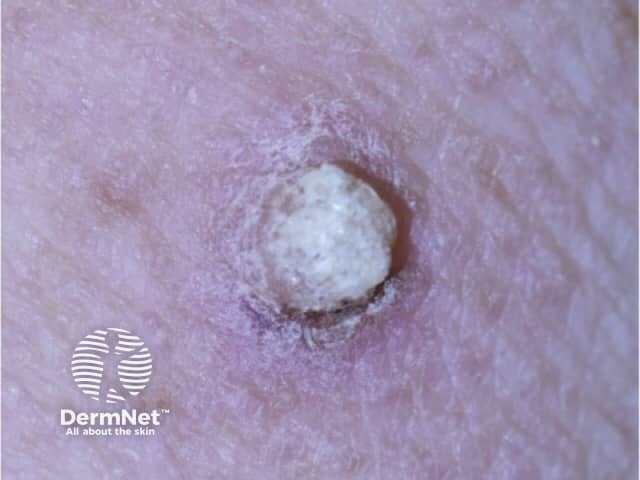
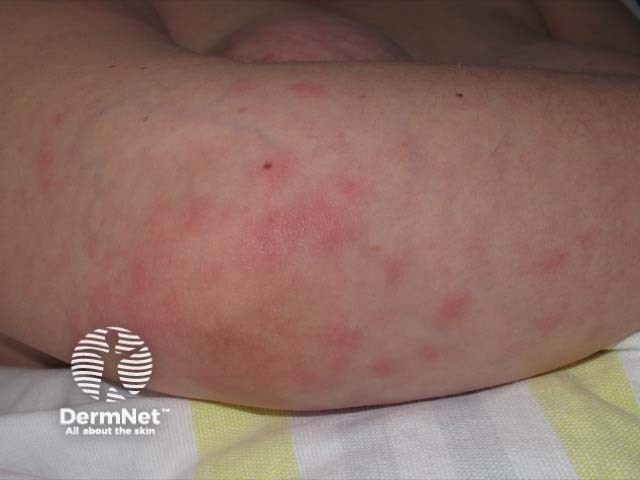
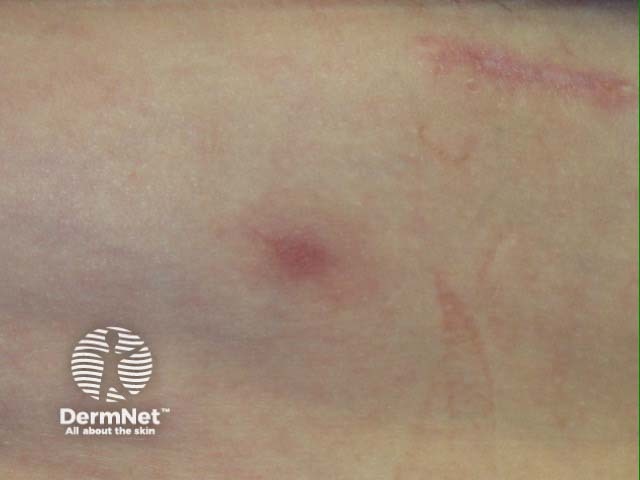
Approved datasheets are the official source of information for medicines, including approved uses, doses, and safety information. Check the individual datasheet in your country for information about medicines.
We suggest you refer to your national drug approval agency such as the Australian Therapeutic Goods Administration (TGA), US Food and Drug Administration (FDA), UK Medicines and Healthcare products regulatory agency (MHRA) / emc, and NZ Medsafe, or a national or state-approved formulary eg, the New Zealand Formulary (NZF) and New Zealand Formulary for Children (NZFC) and the British National Formulary (BNF) and British National Formulary for Children (BNFC).

