Introduction
Demographics
Causes
Clinical features
Variation in skin types
Complications
Diagnosis
Differential diagnoses
Treatment
Outcome
Keratoacanthoma (KA) is a common, rapidly growing, locally destructive skin tumour. KAs may regress spontaneously with scarring, but clinically they may be indistinguishable from well-differentiated squamous cell carcinoma (SCC) and the clinical course may be unpredictable. Thus, many clinicians and pathologists prefer the term SCC, KA-type and recommend surgical excision.
For more images, click here.
Keratoacanthoma is most common in fair-skinned older males with a history of chronic sun exposure. Most patients are over 60 years of age and it is twice as common in males than in females. The incidence rate in Queensland, Australia is 409/100,000 person-years.
There is an increased incidence in:
Keratoacanthoma arises from the infundibulum of the hair follicle. The specific pathogenetic mechanisms are unclear but may involve aberrant regulation of the WNT signal transduction pathways and mutations in the tumour suppression gene TP53.
The risk factors are probably the same as for squamous cell carcinoma, and include:
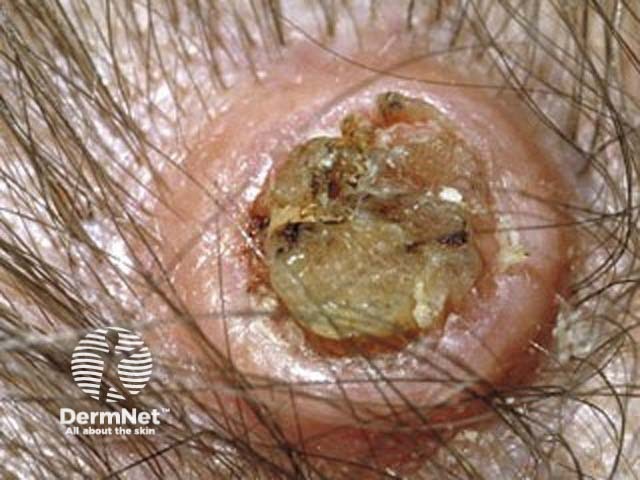
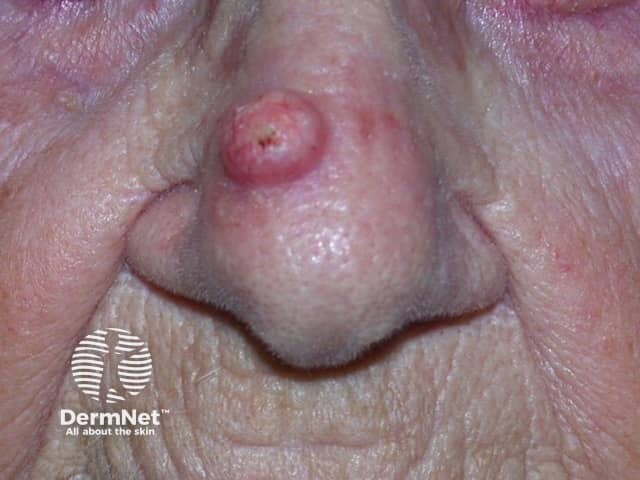
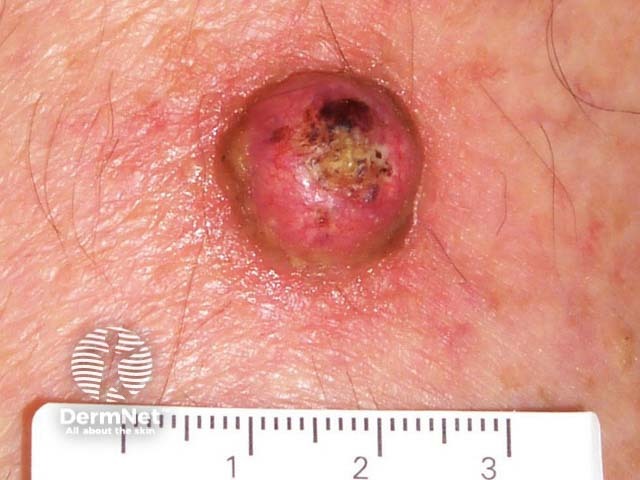
Keratoacanthomas typically present as a solitary, rapidly growing nodule on sun-exposed skin of the face and upper limbs. Keratoacanthomas are sharply demarcated, firm, erythematous or skin-coloured, with a classic central hyperkeratotic plug and an even shoulder. Removal of the keratotic core will leave a ‘crater’-like appearance to the lesion.
There are several variants and syndromes of keratoacanthoma:
Solitary keratoacanthoma (most common)
Keratoacanthoma centrifugum marginatum
Giant keratoacanthoma
Multiple familial keratoacanthoma of Witten and Zak.
Multiple self-healing squamous epithelioma of Ferguson-Smith disease
Keratoacanthomas are much less common in patients with skin of colour, but the clinical features are the same.
The complications of keratoacanthoma include:
Keratoacanthoma is diagnosed on the basis of a typical history, the clinical signs and histopathology.
The classic keratoacanthoma has a crateriform appearance when viewed histologically at low power. Higher power reveals enlarged atypical keratinocytes with eosinophilic cytoplasm that do not extend beyond the level of the sweat glands. These features may be impossible to see in partial or shave biopsy samples, which are not recommended.
Some otherwise typical KAs show squamous cells in a peripheral zone with atypical mitotic figures, hyperchromatic nuclei, and penetration into surrounding tissue. Such lesions are often reported as SCC, KA-type to reflect uncertainty about their true nature.
Unfortunately, dermoscopy cannot reliably discriminate KA from SCC.
Keratoacanthomas must be distinguished from well-differentiated SCC. Other differential diagnoses include:
Most keratoacanthomas are treated surgically. Although they may resolve spontaneously, it is usually prudent to excise them, unless there is clear evidence that regression is in progress.
It is usually best to assume a KA-like lesion is an SCC and to manage accordingly in line with local or national guidance, until proven otherwise.
In selected cases, experienced clinicians may consider other options, such as:
Samples for histology will be absent or may be imperfect, but the above techniques may be deemed suitable after considering the size and location of the tumour, the overall health of the patient and the likely morbidity from surgery.
Keratoacanthoma is regarded as benign and thus has an excellent prognosis following surgical excision.
Lesions that progress and metastasise have probably been SCC, KA-type all along. Patients have an increased incidence of other sun-related skin cancers and should be advised about sun protection and self-examination.

