Introduction Demographics Causes Clinical features Variation in skin types Complications Diagnosis Differential diagnoses Treatment Outcome
Rubinstein-Taybi syndrome (RTS) or broad thumb-hallux syndrome is a rare genetic condition usually caused by mutations of the CREBBP or EP300 genes.
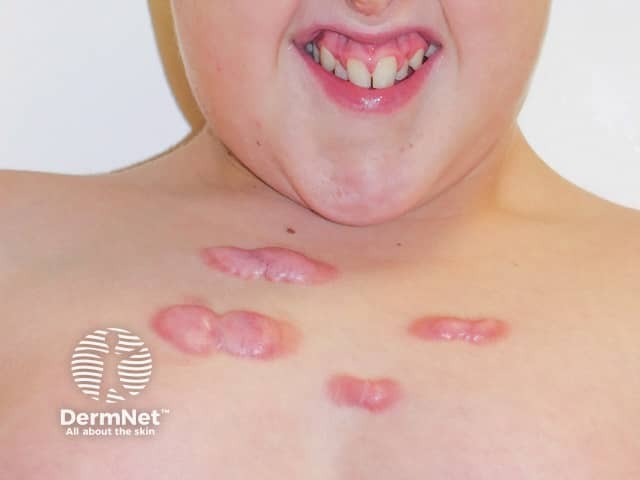
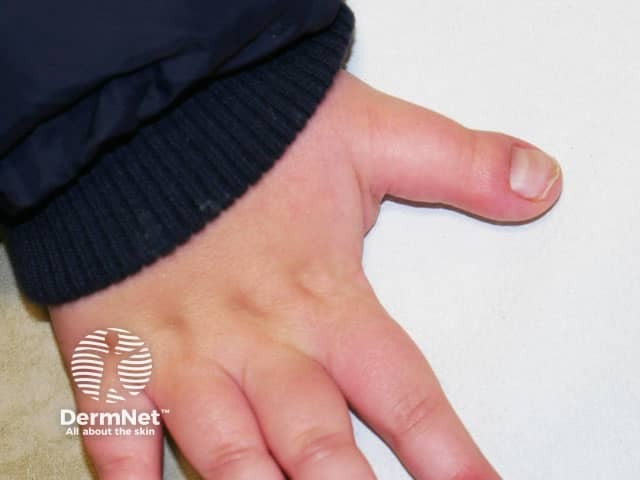
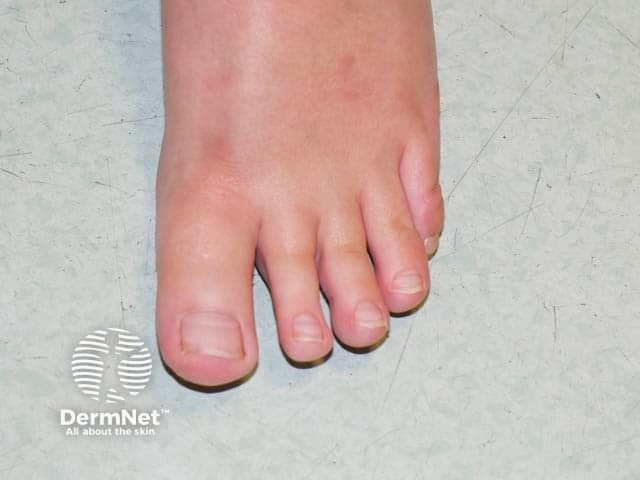
While there is a phenotypic spectrum ranging from mild to severe, RTS is generally characterised by abnormal morphology of the distal phalanges, brachydactyly (short fingers), cardiovascular abnormalities, abnormal dentition and craniofacial abnormalities, developmental delay, and abnormal scarring (principally the presence of atraumatic keloid scarring).
Other names for this condition include RSTS or Rubinstein syndrome.
Named after paediatrician Dr Jack Rubinstein and radiologist Dr Hooshang Taybi, RTS occurs in approximately 1:100,000 to 1:125,000 live births.
Most cases (99%) occur as de novo mutations. However, families with one child diagnosed are often offered genetic counselling due to mosaic presentations. Males and females appear to be equally affected.
RTS is thought to most commonly arise due to microdeletions/ pathogenic variants in the following genes:
CREBBP, found at 16p13.3, encodes a transcriptional co-activator; while EP300 on 22q13 encodes a histone acetyltransferase that along with CREB mediates cAMP gene regulation. Deletions within either of these proteins lead to dysregulation of cell proliferation.
This presents a putative mechanism for explaining many of the features of RST, although the direct pathway between loss of gene expression and the clinical features seen remains elusive. While a de novo mutation causes approximately 99% of cases, these microdeletions can be inherited in an autosomal dominant fashion.
Notably, in many cases, the cause of RTS remains unknown.
While there are no pathognomonic criteria for diagnosis, clinical features include:
Cutaneous symptoms include:
Keloid scars have been described across different skin types, such as case series describing atraumatic keloids in Indian and white populations.
In general, keloid scars appear more common in those with darker skin types, where they often appear less pink and more pigmented.
Rarely diagnosed antenatally, RTS may be identified in the early years of life, often after an investigation in conjunction with testing for other genetic developmental syndromes. Suspicion based on appearance in the clinical examination can be confirmed with genotyping. The presence of a glabellar haemangioma (naevus simplex) in the first weeks of life may be indicative of RTS.
Following diagnosis, consider other investigations to evaluate the extent of the condition and any complications, including a hearing check, ophthalmic exam, echocardiogram, and ultrasound imaging (eg, kidneys, and/or spinal canal if diagnosed in neonatal period).
Diagnosis is usually uncomplicated in patients with the constellation of broad distal phalanges, craniofacial abnormalities, and developmental delay, but there remain a number of differential diagnoses. The differential diagnoses below all have the same typical broad phalanges of RTS but can be differentiated by their unique features:
Further syndromes present with similar craniofacial abnormalities:
There are no specific treatments for RTS. Symptom control depends on an individual’s phenotypic expression. For example, orthopaedic surgery might be considered for significantly angulated or broad halluces or thumbs; while management of ocular or hearing complications may necessitate ophthalmology or audiology expertise.
Serial review is encouraged for patients diagnosed with RTS for early detection of cardiac, renal, ophthalmic, dental, ENT, and other abnormalities. For example, at least annual ophthalmic and hearing checks are suggested, along with regular growth monitoring, and a dental check-up approximately every 6 months.
RTS-associated keloid scars appear to respond poorly to existing treatment regimens and further work is required to identify optimal treatments.
Investigation into epidrugs (medications that modify the expression of individual genes) is confined to a few predominantly laboratory-based trials, but has shown early promise as a future therapy for genetic diseases including RTS.
Despite the reported increased incidence of benign tumours, life expectancy in people with RTS seems to be comparable to that of the unaffected population.
Developmental and motor delays may require long-term support, and regular monitoring for complications is recommended.

