Spitz naevus Spitzoid tumour of uncertain significance, atypical Spitz naevus, Spitzoid melanoma Worrisome features in Spitz naevi Pigmented spindle cell naevus of Reed Differential diagnosis Spitz tumours with kinase fusions
Spitz naevus may pose great diagnostic difficulty due to clinical, dermatoscopic and histologic features resembling melanoma. Though usually seen in children, Spitz naevi may arise in adults. The typical or classic lesion is a pink papule or nodule, which appears suddenly and grows rapidly for a few months. There is a pigmented variant.
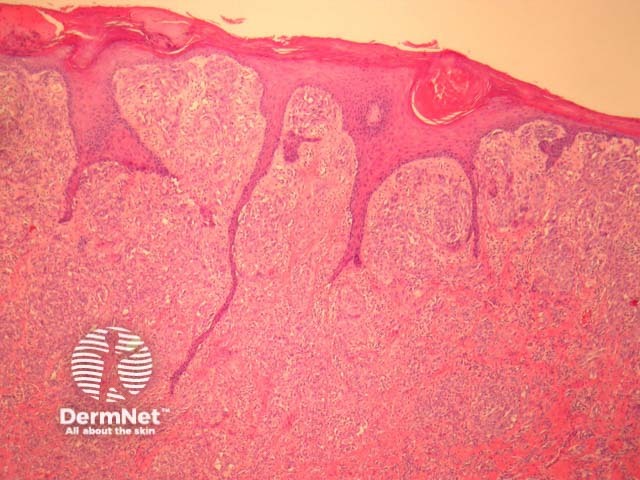
The majority of Spitz naevi are compound or intradermal tumours. Purely junctional Spitz naevi are fortunately quite rare as they can closely resemble melanoma. Various histologic diagnostic criteria exist, which can help differentiate Spitz naevi from melanoma.
Major diagnostic criteria for Spitz naevi:
Minor diagnostic criteria for Spitz naevi include junctional cleavage, superficial multinucleate naevus cells, a fibrovascular stroma, and solitary naevus cells at the base of the lesion.
Innumerable variants of Spitz naevi have been described. They have little clinical relevance but may be highly relevant for junior faculty seeking publication and promotion.
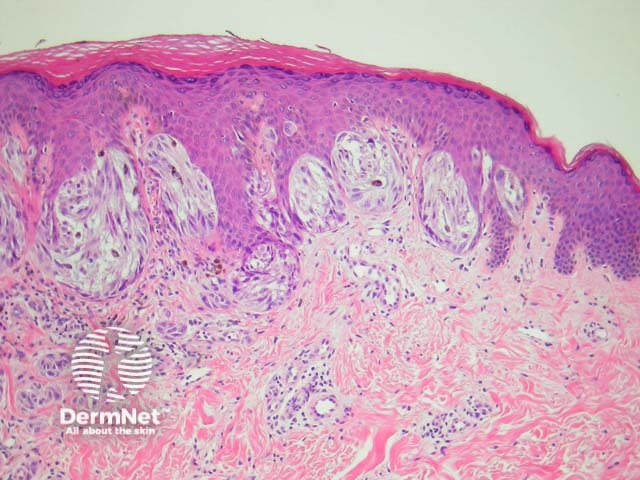
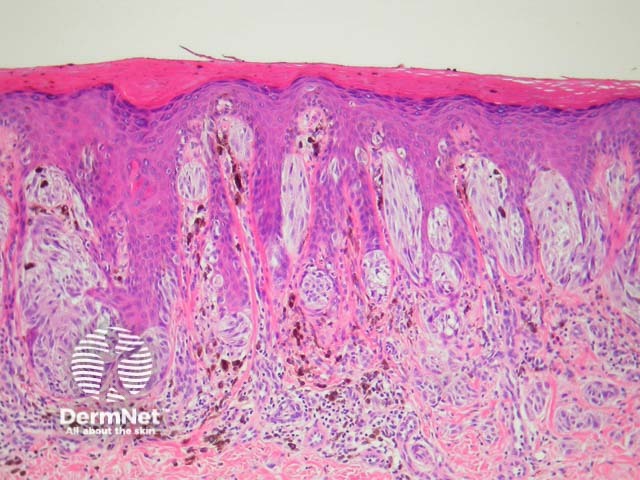
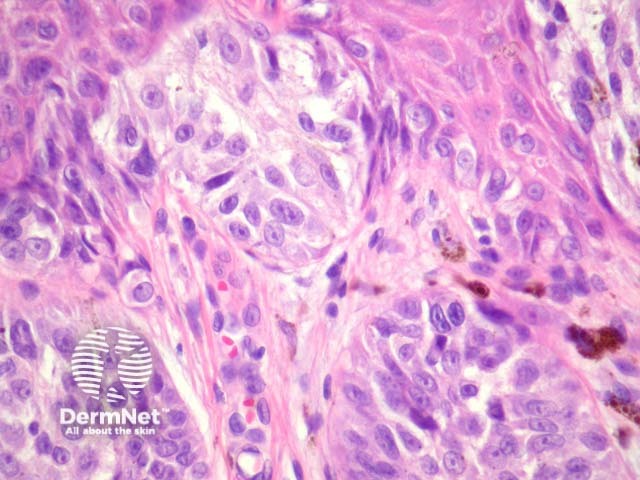
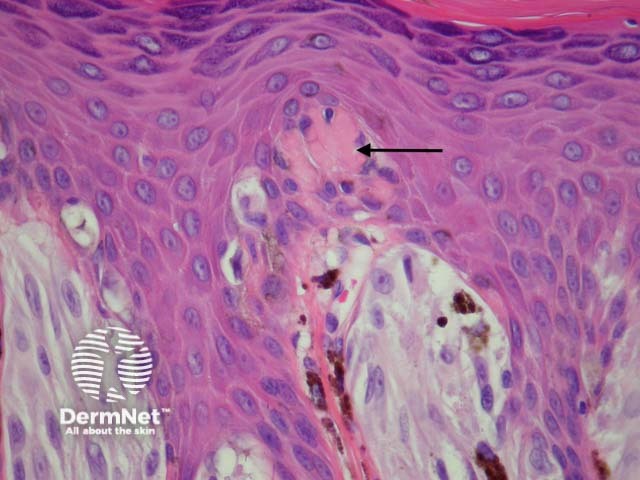
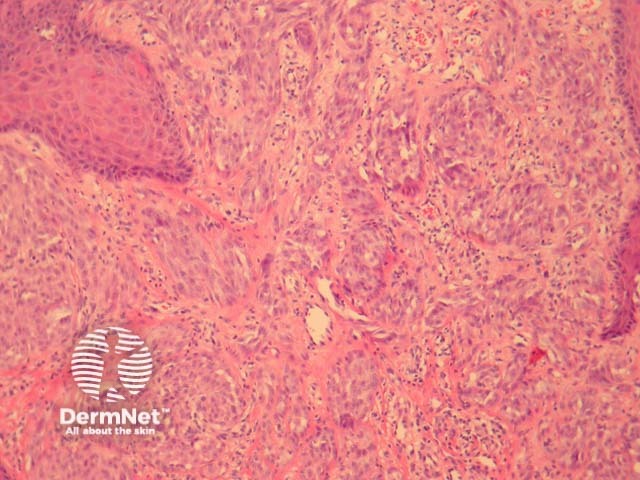
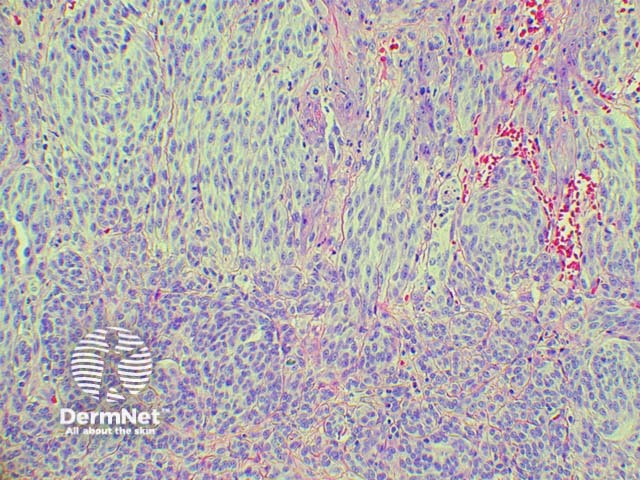
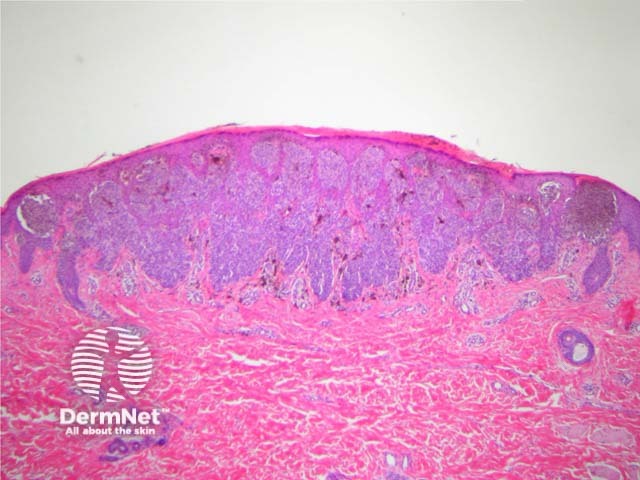
It has become increasingly clear that a Spitzoid Tumour of Uncertain Significance or STUMP represents a tumour of intermediate malignant potential, midway in a histological continuum with Spitz naevus at the benign end and spitzoid melanoma at the malignant end. STUMP quite frequently involve lymph nodes but seldom spreads beyond (figure 5).
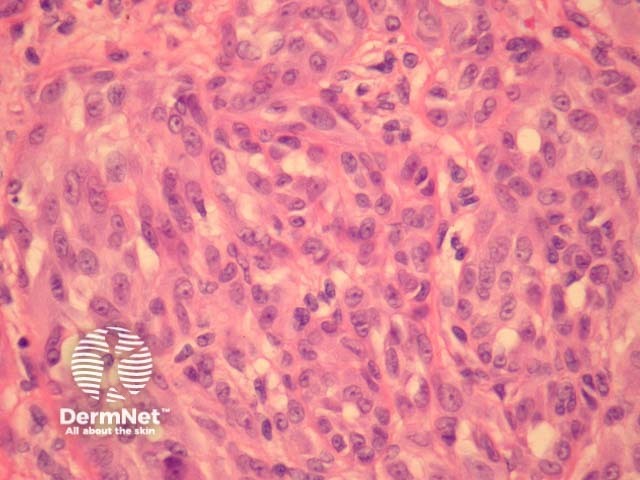
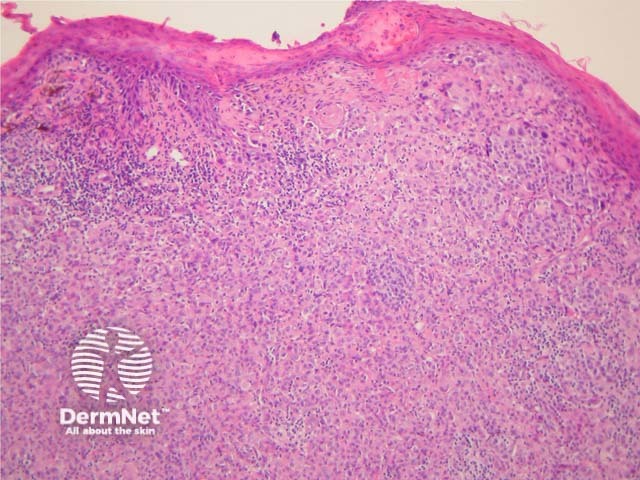
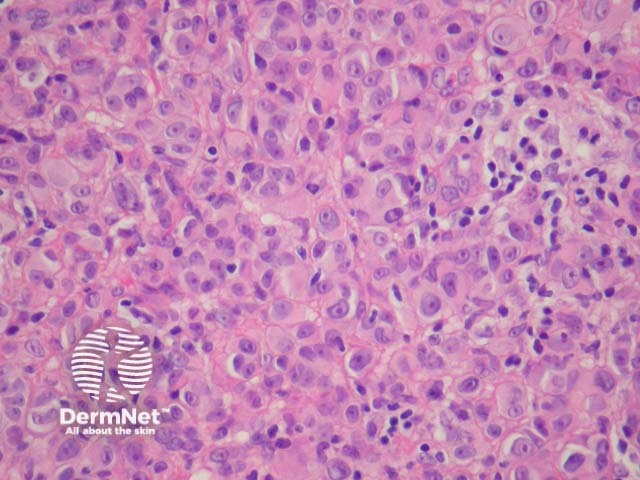
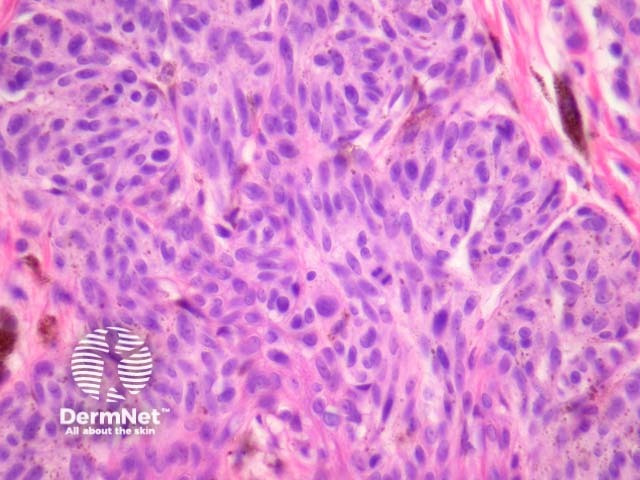
Malignant Spitz tumours are rare but potentially lethal. They are generally very atypical (figure 6, 7), but, unfortunately, some of these malignant cases have features which almost precisely mimic a benign Spitz naevus. In approaching Spitzoid lesions, the concerning histologic features are listed in the table below. Unfortunately, none of these features provides absolute certainty about the diagnosis (with the possible exception of a 9p21 loss by FISH)
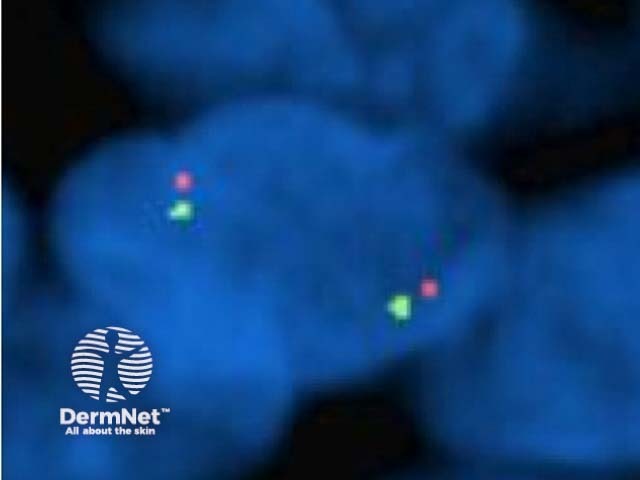
Comparative Genomic Hybridisation (CGH) can be extremely useful in difficult cases but funding for these tests can be problematic. In New Zealand, FISH is currently available privately through IGENZ laboratory in Auckland. Figure 8 shows a normal diploid complement of 9p21 (ie two copies of each colour) in each cell, which is more consistent with a Spitz naevus with a low malignant potential.
Pigmented spindle cell naevus of Reed appears in children and young adults as a well-defined heavily pigmented lesion with a rather characteristic dermatoscopic star-shaped morphology. They may be difficult to distinguish from pigmented Spitz naevi but distinction may not be a real issue as they likely variants of the same entity and may co-exist.
Histologically, there are highly characteristic nests of spindle-shaped cells with heavy pigmentation. Kamino bodies and junctional clefting (as seen in Spitz naevi) may be present. Pagetoid spread may be present but should be mainly limited to the lower epidermis or if higher than this only seen in the middle of the naevus (figures 9, 10).
The differential diagnosis of Spitz and Reed naevi includes acquired melanocytic naevi, blue naevi and melanoma.
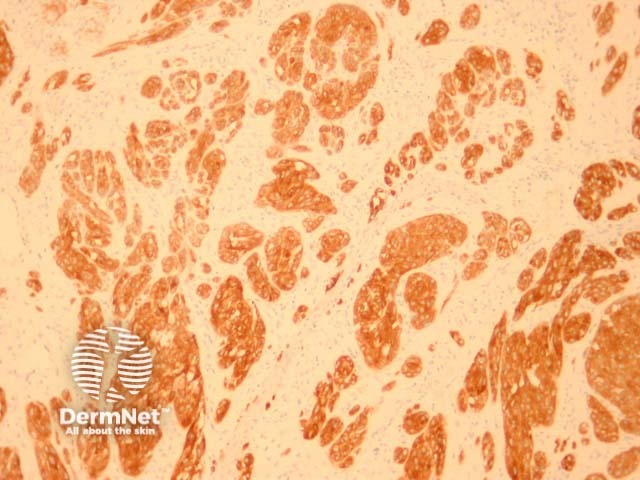
It has recently been shown that spitzoid neoplasms harbour kinase fusions of ROS1 (17%), NTRK1 (16%), ALK (10%), BRAF (5%) and RET (3%) in a mutually exclusive pattern. These mutations appear to be present in a subset of both benign and malignant Spitzoid tumours. ALK-mutated Spitz tumours are often compound melanocytic proliferations with a predominant intradermal growth. These lesions often have a plexiform dermal growth of intersecting fascicles of fusiform melanocytes (figures 11, 12, 13). ALK immunohistochemistry can confirm this fusion (figure 14). Some of these mutations may provide therapeutic targets for malignant tumours.