Introduction
Demographics
Causes
Clinical features
Complications
Diagnosis
Differential diagnoses
Treatment
Outcome
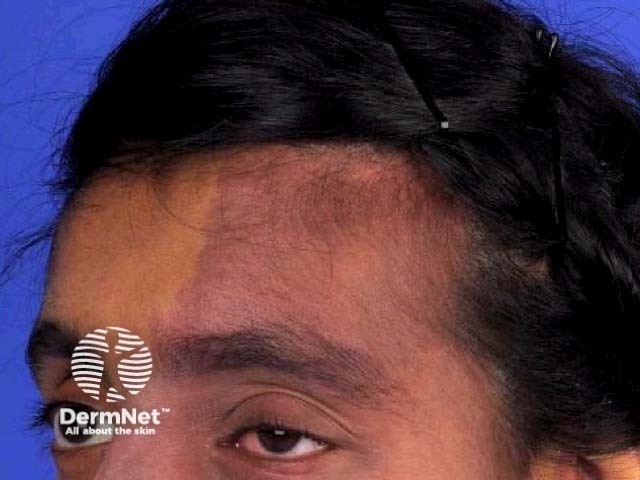
Sturge–Weber syndrome is a rare, congenital, and non-inherited neurocutaneous disorder characterised by capillary malformation on the facial skin (port-wine stain) and capillary-venous malformations in the brain and in the eyes [1].
It is estimated that approximately one in every 20,000 to 50,000 babies are born with Sturge–Weber syndrome [2]. As it is caused by a somatic mosaic mutation, it is not inherited from the parents.
Sturge–Weber syndrome is caused by a somatic mosaic mutation of the GNAQ gene on chromosome 9 [3]. This means that the mutation in the gene has occurred in the body cells after the formation of the zygote. GNAQ regulates intracellular signalling pathways. The mutation gives rise to uncontrolled formation or maturation of capillaries in affected cells.
Sturge–Weber syndrome is characterised by vascular malformations on the face and in the eye and brain of affected individuals. These are present at birth.
Neurological and ophthalmological signs in Sturge-Weber syndrome are progressive and usually develop in the first two years of life. These can include:
The complications of Sturge–Weber syndrome depend on the extent of vascular malformations and other clinical features. They are extremely variable.
The diagnosis of Sturge–Weber syndrome is based on finding the characteristic trigeminal port-wine stains and leptomeningeal capillary-venous malformations. A diagnosis based on leptomeningeal lesions alone depends on the development of symptoms.
If there are neurological symptoms or findings, magnetic resonance imaging (MRI) of the brain is undertaken with gadolinium contrast to detect leptomeningeal capillary-venous malformations.
The characteristics of other vascular malformation syndromes are described below.
There is no specific treatment for Sturge–Weber syndrome. Treatment consists of managing the cutaneous, neurological and ocular symptoms, with limited success.
Treatment of seizures in patients with Sturge–Weber syndrome with antiepileptics is not always successful [8]. No single treatment appears to be superior to others.
Port-wine stains can be treated with pulsed-dye laser with variable results [9]. Due to the generally poor results of pulsed-dye lasers alone, topical antiangiogenic agents are being trialled as adjunctive therapies.
As glaucoma is a common complication of Sturge-Weber syndrome, regular eye checks with an ophthalmologist are recommended. Glaucoma is treated surgically and medically [10].
Infants diagnosed with Sturge–Weber syndrome should be treated with low-dose aspirin [11,12]. The antithrombotic therapy may prevent the progression of the disease, which can impair blood flow to the brain resulting in neuronal damage.
The prognosis of Sturge–Weber syndrome depends on the extent of involvement of the brain and the skin. Extensive port-wine stains are associated with a higher risk of epilepsy and glaucoma, while bilateral leptomeningeal vascular malformations are associated with learning and intellectual disability [13]. The onset of seizures before the age of one has a significant effect on cognitive and motor function in children with Sturge–Weber syndrome [14].
It is estimated that approximately one in two adults with Sturge–Weber syndrome have neurological defects, even in those who were initially asymptomatic.

