Introduction
Demographics
Causes
Clinical features
Complications
Diagnosis
Differential diagnoses
Treatment
Outcome
VEXAS (Vacuoles, E1 enzyme, X-linked, Autoinflammatory, Somatic) syndrome is an adult-onset severe inflammatory and/or haematological disorder due to a somatic mutation in the UBA1 gene on the X chromosome, first described in 2020.
Although initially thought to be rare, since the first description of VEXAS syndrome in October 2020 it is being increasingly diagnosed on retrospective genetic testing of patient cohorts identified by clinical features including series from France, the Netherlands, and the USA, as well as individual case reports.
VEXAS syndrome is almost exclusively described in males, with most cases diagnosed in mid to late adult life. At least 70 cases have now been reported in recent months with an age range of 45–80 years and a median age at symptom-onset in the late 50s to mid 60s.
There has been one female with VEXAS syndrome reported who had a single X-chromosome.
VEXAS syndrome is due to an acquired somatic (mosaic or postzygotic) mutation affecting the methionine-41 codon (p.Met41) in UBA1. This gene encodes the major E1 enzyme involved in the activation of ubiquitin, a small regulatory protein which attaches to substrate proteins (ubiquitylation). The p.Met41 mutation results in decreased ubiquitylation particularly in haematopoeitic stem cells with subsequent activation of the innate immune system.
VEXAS syndrome is clinically heterogeneous but is characterised by autoinflammatory, treatment-resistant manifestations most commonly involving the skin and bone marrow.
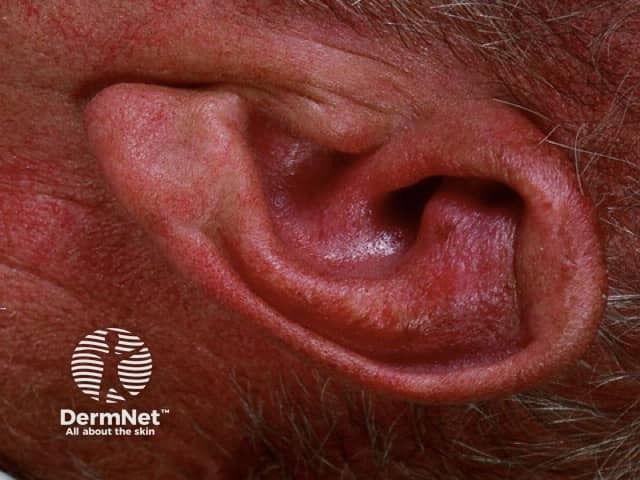
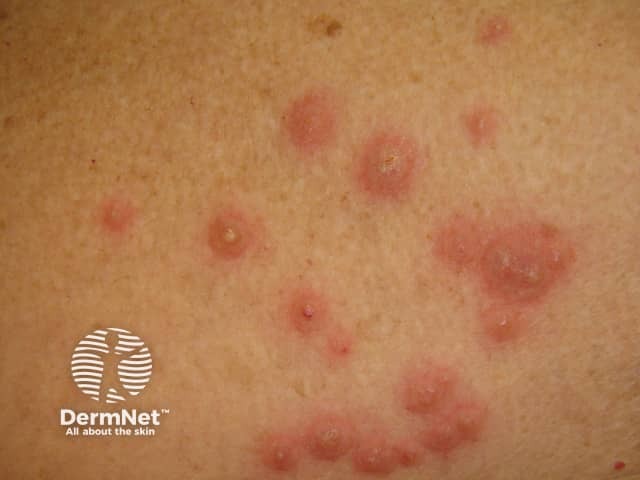
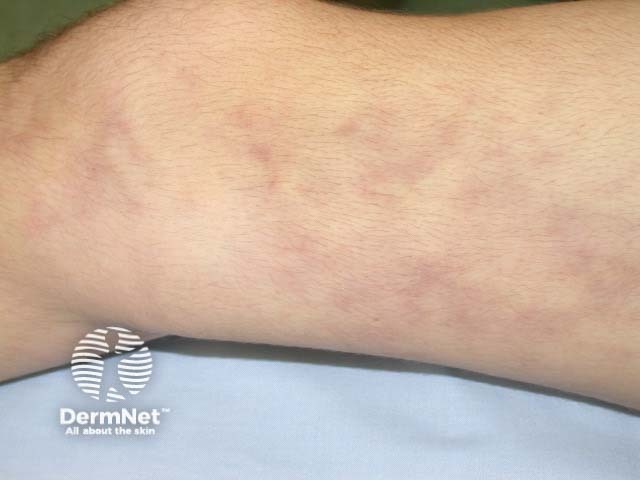
VEXAS syndrome should be considered in the male patient with complex overlapping inflammatory and haematological features, although all components may not be present at initial presentation.
Investigations may include:
VEXAS syndrome presenting with dermatological manifestations may be initially diagnosed as having the specific dermatosis.
VEXAS syndrome typically responds poorly to treatment, with high dose systemic corticosteroids (> 20mg/d) showing the most consistent benefit for the inflammatory features. Other DMARDs and immunosuppressive drugs are ineffective. Colchicine may reduce the steroid requirement. Tocilizumab is a theoretical option being trialled.
Treatment for myelodysplastic syndrome, multiple myeloma, or MGUS have been mostly unsuccessful with a partial response at best. Autologous stem cell transplantation in one patient induced only a short-lived remission of 6 months before symptoms recurred.
VEXAS syndrome is a chronic progressive autoinflammatory syndrome. Patients have often had symptoms for 4-5 years before the diagnosis is made. However, the long-term prognosis appears poor: the mortality rate in a Netherlands study was 50% and in a series from the USA, 9/16 died from disease-related causes.

