Introduction
Demographics
Causes
Clinical features
Diagnosis
Treatment
Outcome
A café-au-lait macule is a common birthmark, presenting as a hyperpigmented skin patch with a sharp border and diameter of > 0.5 cm. It is also known as circumscribed café-au-lait hypermelanosis, von Recklinghausen spot, or abbreviated as 'CALM'.
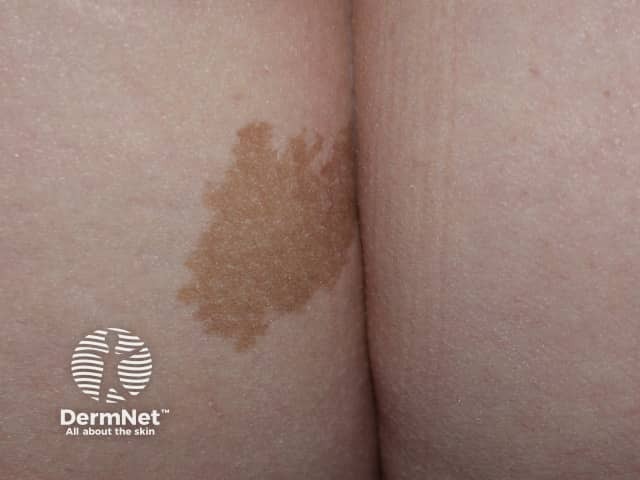
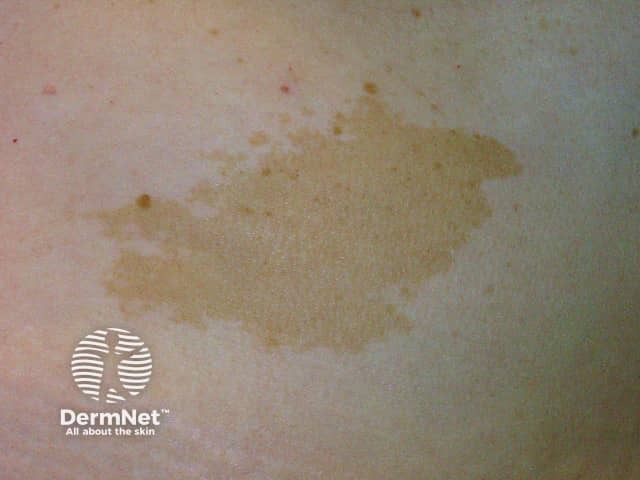
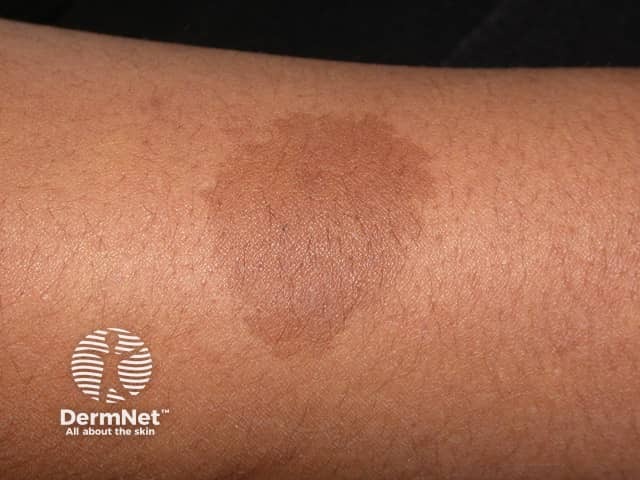
See more images of café-au-lait macules.
Café-au-lait macules are usually present at birth (congenital) or appear in early infancy. They sometimes become apparent later in infancy, especially after exposure to the sun, which darkens the colour.
They may be isolated or associated with systemic diseases such as neurofibromatosis (NF), McCune Albright syndrome, Legius syndrome, and Noonan syndrome with multiple lentigines syndrome.
The overall prevalence of café-au-lait macules varies with race:
Isolated café-au-lait macules are invariably solitary. More than 3 in a Caucasian or more than 5 in an African American are uncommon and should lead to systemic evaluation, referral, and close follow-up.
The brown colour of a café-au-lait macule is due to a pigment called melanin, which is produced in the skin by cells called melanocytes.
Multiple café-au-lait macules are related to several genetic syndromes.
About half of those with neurofibromatosis type 1 (NF1) have an inherited mutation of the NF1 gene on chromosome 17. NF1 codes for neurofibromin, a tumour suppressor gene. Others have a sporadic mutation of the same gene.
Like NF1, autosomal dominant and sporadic mutations of the NF2 gene are equally common. NF2 gene codes for Merlin protein, whose physiologic function is still under investigation.
Legius syndrome is caused by SPRED gene mutation, which generally controls the RAS pathway and interacts with neurofibromin.
McCune Albright syndrome is caused by mutation of Gs protein, activating adenylate cyclase.
Noonan syndrome with multiple lentigines is due to the autosomal dominant inheritance of mutated PTPN11 gene on chromosome 12. The gene codes protein tyrosine phosphatase SHP-2.
The next three syndromes are much rarer than those described above.
Watson syndrome is linked to mutation of NF1 gene, or is at least allelic to NF1, or is caused by mutation of contiguous genes to NF1.
Bloom syndrome is due to the autosomal recessive mutation in BLM gene on chromosome 15. The gene product is DNA helicase, an enzyme essential to DNA repair to prevent chromosomal breakage.
There are several genetic abnormalities associated with Silver-Russell syndrome. The two most common abnormalities are:
Café-au-lait macules:
The distribution and configuration of café-au-lait macules can be a clue to an underlying syndrome.
NF1 is highly variable in appearance. The main 2 National Institute of Health (NIH) Consensus criteria for the diagnosis of NF1 are:
There are 5 other NIH criteria.
At least two criteria are required to make a working diagnosis of neurofibromatosis. The definitive diagnosis is made by confirming the presence of a genetic mutation.
NF2 presents with unilateral or bilateral acoustic schwannoma (vestibular schwannoma). Patients develop hearing problems, ringing in the ears (tinnitus), and dizziness. By the age of 30, nearly all patients with NF2 have bilateral vestibular schwannoma.
Patients with Legius syndrome have multiple café-au-lait macules (> 5mm in children and > 15 mm in adults). Axillary freckling less common.
They may rarely have macrocephaly, cognitive disabilities, and several congenital malformations such as Noonan-like facies, pectus excavatum/carinatum, and lipomas.
Café-au-lait macules in McCune Albright syndrome are fewer than in NF1, with more irregular borders. They are classically found on the midline.
The clinical diagnosis of McCune Albright syndrome is established by a triad of abnormalities:
Noonan syndrome with multiple lentigines is also known as LEOPARD syndrome; the L of LEOPARD syndrome refers to prominent lentigines. These are multiple < 5 mm, well-demarcated, brown macules presenting on the whole skin without mucous membrane involvement. They appear at birth and continue increasing in number until puberty.
LEOPARD is an acronym referring to the clinical findings required to make the diagnosis:
Patients with Noonan syndrome with multiple lentigines may also develop café-au-lait macules, nail malformation, and hyperelastic skin.
Watson syndrome is extremely rare with only 4 families described between the 1960s to early 1990s. Their café-au-lait macules in Watson syndrome had similar characteristics to NF1. Other features of Watson syndrome are:
Café-au-lait macules are not the main clinical finding in Bloom syndrome. Key characteristics of Bloom syndrome are:
Even though café-au-lait macules are not essential for diagnosis, they are common in children with Silver-Russell syndrome. They are mainly located on chest, stomach, and extremities.
The main features of Silver-Russell syndrome are:
Café-au-lait macules are diagnosed clinically. If significant in number and size, a complete clinical examination should be undertaken to determine whether an associated syndrome may be present.
Syndromes may be diagnosed from their clinical manifestations or by genetic testing.
No medical care is required to treat café-au-lait macules. Lasers reported to have successfully faded café-au-lait macules include:
Results are inconsistent. One group has found lesions with an irregular margin respond better than those with a smooth, well-defined border. Risks for laser surgery include transient/permanent hyperpigmentation, hypopigmentation, and scarring.
Treatment of underlying syndromes may be complex and require multidisciplinary care.
Without treatment, café-au-lait macules persist lifelong. Results from lasers are not consistent. However, for those who responded to initial treatment, recurrence rates are reported to be low.

