Introduction Demographics Causes Clinical features Diagnosis Differential diagnoses Treatment Outcome
Legius syndrome is a rare genetic disorder that was first described in 2007 [1]. It is also known as neurofibromatosis type 1-like syndrome [2].
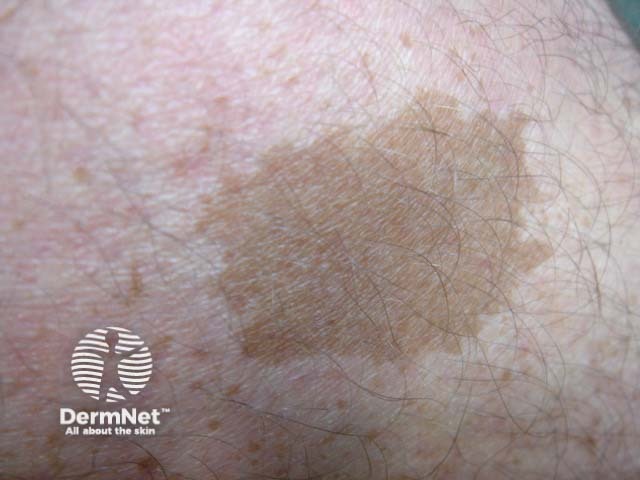
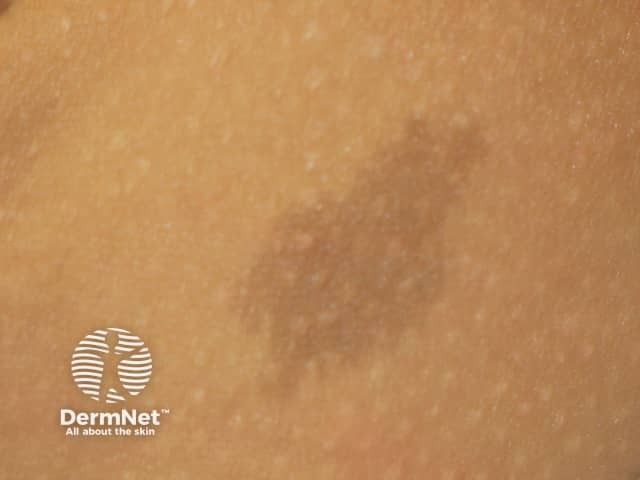
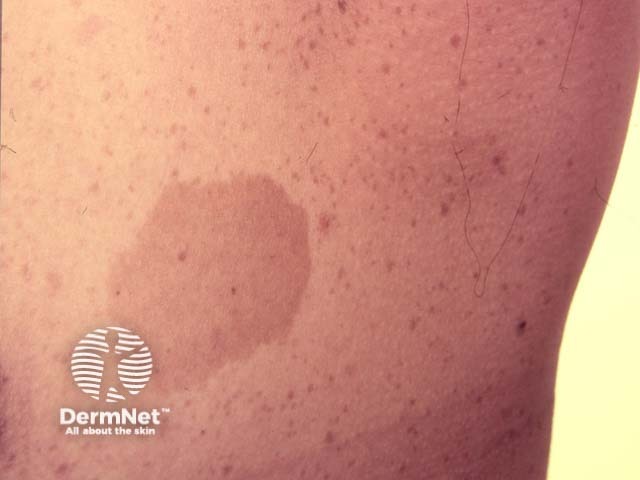
Legius syndrome is classically characterised by multiple light-brown macules, known as café-au-lait macules [3]. Unlike neurofibromatosis type 1, there are no tumours found in Legius syndrome.
Legius syndrome may occur in anyone who has at least one biological parent with genetically confirmed Legius syndrome [2]. The prevalence of Legius syndrome is unknown, but it may be higher than expected due to misdiagnosis as neurofibromatosis type 1 [4].
Legius syndrome is a genetic condition inherited in an autosomal dominant manner that involves a SPRED1 gene mutation on chromosome 15q14. This mutation results in the abnormal function of the SPRED1 protein, which is responsible for regulating specific cell-signalling pathways involved in cell proliferation, differentiation, and apoptosis. No other pathogenic variants have been found to cause Legius syndrome [2,3].
As an autosomal dominant condition, Legius syndrome may occur in anyone with at least one biological parent with genetically confirmed Legius syndrome. Rarely, it may also occur de novo, but these cases have not been evaluated sufficiently to confirm this [2]. Each child with a parent with genetically confirmed Legius syndrome has a 50% risk of inheriting the condition.
The clinical features of Legius syndrome vary significantly in nature and severity from one person to another.
Almost all patients with Legius syndrome present with multiple café-au-lait macules [2,3]. The number of these macules tends to increase during childhood [4].
Other common cutaneous features may include:
Non-cutaneous features may include:
Neuropsychiatric features may include:
The intellectual disabilities are generally less severe in patients with Legius syndrome compared to patients with neurofibromatosis type 1 [5].
Importantly, Legius syndrome does not cause neurofibromas and Lisch nodules, which are typically seen in neurofibromatosis type 1 [2,3].
Legius syndrome is difficult to diagnose on a clinical basis alone given its similar cutaneous clinical presentation to other disorders with multiple café-du-lait macules.
Where Legius syndrome is suspected, genetic testing can be used to confirm the diagnosis. This involves:
Suspicious findings that may warrant genetic testing for Legius syndrome include:
Legius syndrome is often misdiagnosed because its pigmentary manifestations are very similar to those seen in other syndromes with multiple lentigines. Correct diagnosis is critical to guide long-term monitoring and management.
Other syndromes characterised by multiple lentigines include:
Children with Legius syndrome should be under regular screening and evaluation for developmental delay, cognitive impairment, and behavioural issues [2].
The treatment of Legius syndrome is primarily supportive and should be focused on the specific problems of the affected individual.
Appropriate management, if indicated, may include:
Patients with Legius syndrome have a generally good prognosis with appropriate problem-based management.
An affected individual must consider the 50% risk of genetic transmission to each child.

