

Erythropoietic protoporphyria — extra information
Introduction - porphyrias
Introduction - erythropoietic protoporphyria
Demographics
Causes
Clinical features
Diagnosis
Treatment
What are the porphyrias?
The porphyrias are a group of genetic diseases in which there are enzyme deficiencies in the haem pathway; haem is the part of haemoglobin that gives blood its red colour. An enzyme deficiency results in a build-up of precursors in the blood and other organs. The precursor chemicals can cause acute attacks in the acute hepatic porphyrias and photosensitivity and skin lesions in the cutaneous porphyrias.
What is erythropoietic protoporphyria?
Erythropoietic protoporphyria (EPP) is one of the cutaneous porphyrias. EPP is due to an inherited deficiency of the enzyme ferrochelatase. Reduced activity of this enzyme causes a build-up of protoporphyrin in the skin resulting in photosensitivity.
Abnormally high levels of protoporphyrin can rarely cause liver disease.
Who gets erythropoietic protoporphyria?
Erythropoietic protoporphyria (EPP) is the second most common of the skin porphyrias. It occurs in approximately 1:100 000 of the population. It is inherited in the vast majority of patients, but most will not have a family history of the disease as it is not common to be passed down; the risk of developing EPP is approximately 1 in 10 to the offspring of an affected parent. Males and females are equally affected.
First symptoms usually appear in infancy or early childhood
What causes erythropoietic protoporphyria?
EPP is thought to be due to a compound loss-of-function mutation in the gene encoding ferrochelatase (FECH; 612386) found on chromosome 18q21. Typically, there is a mutation on one gene as well as a second, low-expression allele. Rarely, there is a gain in function mutation in ALA synthase (ALAS)-2 (X-linked dominant inheritance). Thus, inheritance can be autosomal recessive or autosomal dominant with incomplete penetrance (96% of patients in the UK).
In very rare instances, EPP has been reported to have been caused by myelodysplasia or myeloid leukaemia due to associated chromosomal instability, including knock-out loss of chromosome 18.
What are the clinical features of erythropoietic protoporphyria?
Cutaneous symptoms and signs of EPP
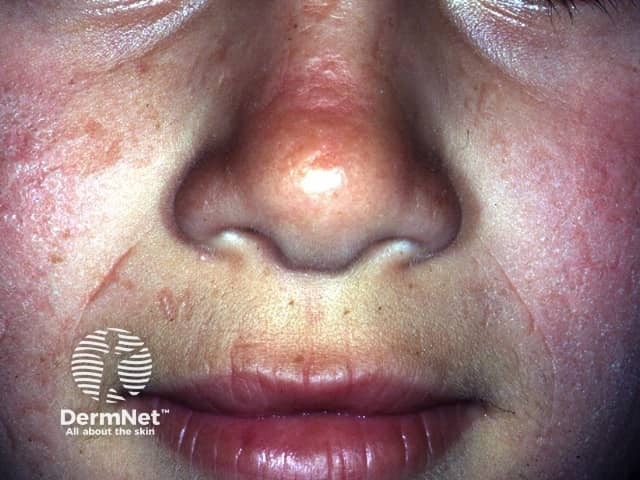
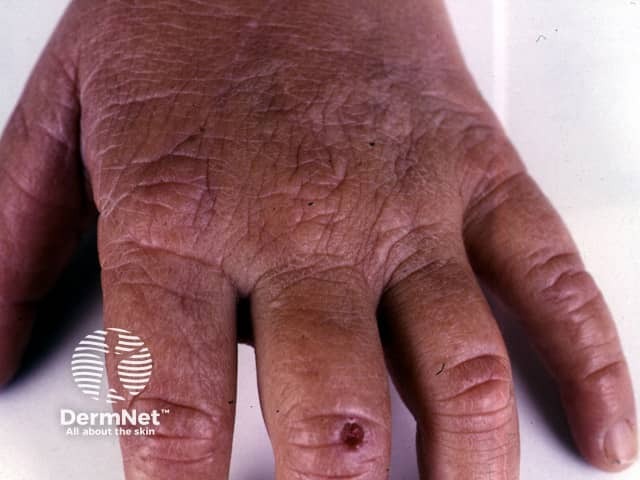
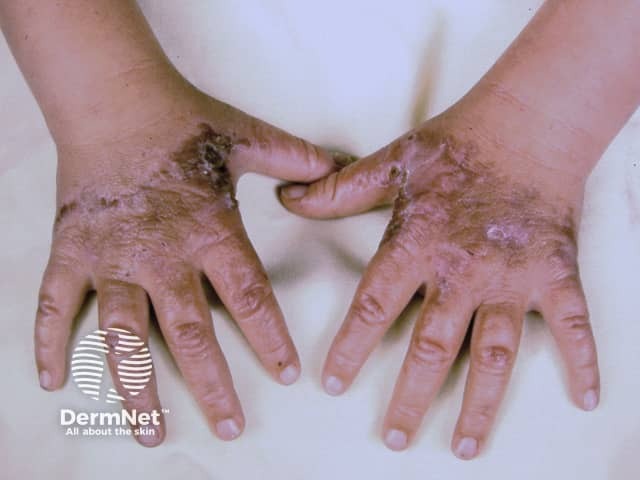
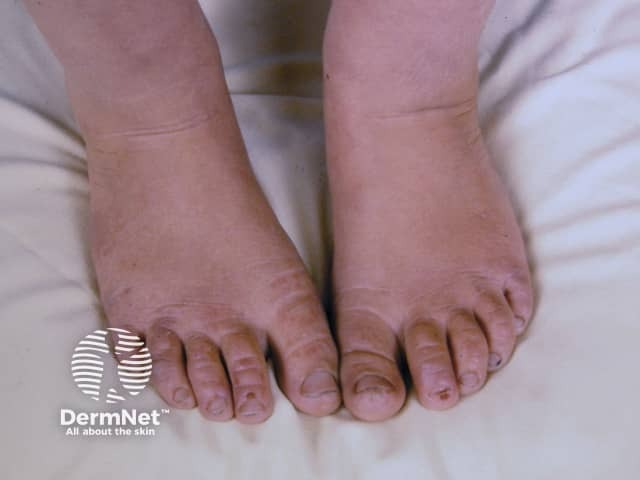
EPP causes skin pain on exposure to sunlight, most often on the tops of the hands and feet, face and ears. Pain can be severe and last days after sun exposure. There may not be anything to see at the time. Prolonged exposure can result in some redness and swelling, and uncommonly in blistering and crusting.
- EPP can be difficult to diagnose in childhood — babies may cry in the sun or in the car.
- Symptoms are worse in summer and in sunny climates.
- Over the years, the skin on the backs of the hands and cheeks can have some thickening with subtle pitted scarring.
- Severity can range from mild, where hours of sun are required to provoke pain, to severe, where the onset of pain is within seconds.
- In most cases visible changes are mild.

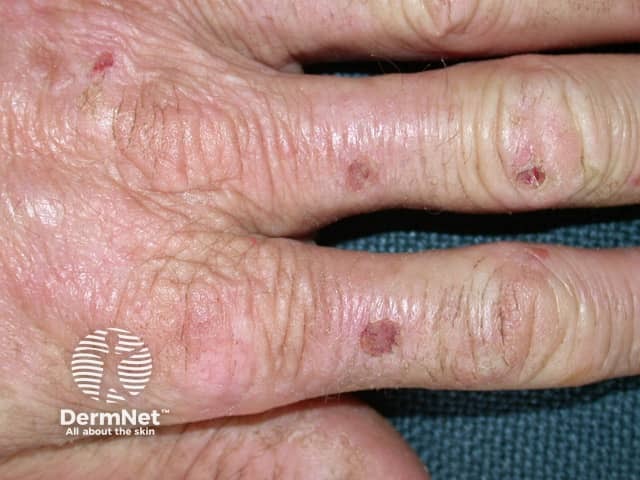
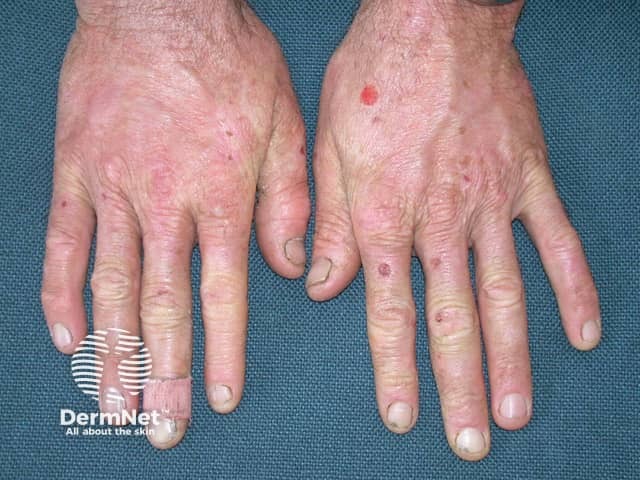
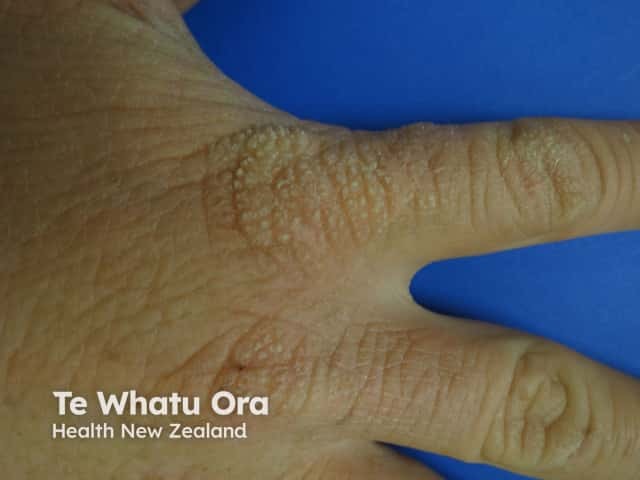
Liver disease in EPP
People with EPP-induced liver disease often have mild changes in liver blood tests. About 10% develop more severe liver disease, presenting with malaise, right upper abdominal pain, jaundice and eventually, liver failure (with increasing sun sensitivity).
Gallstones are common in patients with EPP.
How is erythropoietic protoporphyria diagnosed?
A clinical diagnosis of EPP is often made during childhood. The blood needs to be sent for porphyrin analysis in a tube protected from light with aluminium foil.
- The patient's red blood cells may be noted to fluoresce by ultraviolet microscopy.
- The characteristic change is the elevation of the red cell protoporphyrin.
- Genetic testing for mutations in the ferrochelatase (FECH) gene can be performed.
- A skin biopsy is rarely performed as the skin often appears normal; however, EPP has some characteristic features on histopathology.
Once the diagnosis has been made, regular checks of liver function are required and intermittent liver imaging.
What is the treatment for erythropoietic protoporphyria?
There is no cure for EPP.
Lifelong photosensitivity is the main problem.
- Once the pain has started, pain relief can be difficult to achieve. Most patients immerse the affected areas in cold water or use ice packs. Topical anaesthetic creams can be helpful.
- To reduce pain, avoid unnecessary exposure to sunlight and wear protective clothing and wide-brimmed hats.
- Car windows should be tinted.
- Other sources of light may also cause symptoms, including fluorescent and halogen lights.
- Protect the skin from exposure to operating lamps during a surgical procedure.
- Sunscreens may be helpful, especially formulations containing zinc oxide or titanium dioxide that reflect visible light.
- Vitamin D supplementation is appropriate in patients that strictly avoid exposure to sunlight and in those on systemic photoprotection such as afamelanotide.
Avoidance of alcohol is important to reduce the risk of liver damage and liver failure.
Trials of treatment for EPP have been difficult to assess. Effective treatment should reduce pain and increase time outdoors without pain.
- Narrowband UVB phototherapy does not cause the EPP pain. It is given 3 days a week over 6 weeks in Spring. It increases melanin content (causing a tan) and induces skin thickening so to provide some level of protection from the sun.
- Oral antioxidants such as beta-carotene, Polypodium leucotomas extract, warfarin and N-acetyl cysteine have been used but studies showing effectiveness are lacking.
- Iron supplementation should be avoided — unless severely iron deficient — as iron can increase photosensitivity in EPP.
- Afamelanotide, an α-melanocyte stimulating hormone given by subcutaneous implantation, has been reported to provide clinical effectiveness and safety in EPP. It is approved by the European Medicines Agency and in the USA for the treatment of EPP under orphan drug status.
- Patients with EPP that also have liver disease require specialist medical treatment and possibly liver transplantation.
Another more serious condition, congenital erythropoietic porphyria, is now curable by stem cell transfusion, paving hope for the future, but there is not yet a cure available for EPP.
References
- OMIM – Online Mendelian Inheritance in Man (search term Erythropoietic protoporphyria)
- Biolcati G, Marchesini E, Sorge F, Barbieri L, Schneider-Yin X, Minder EI. Long-term observational study of afamelanotide in 115 patients with erythropoietic protoporphyria. Br J Dermatol 2014; 172: 1601–12. DOI: 10.1111/bjd.13598. PubMed
- Fabrikant J, Touloei K, Brown SM. A review and update on melanocyte stimulating hormone therapy: afamelanotide. J Drugs Dermatol 2013; 12: 775–9. Review. PubMed
- Book: Textbook of Dermatology. Ed Rook A, Wilkinson DS, Ebling FJB, Champion RH, Burton JL. Sixth edition. Blackwell Scientific Publications.
- Falchetto R. The Patient Perspective: A Matter of Minutes. Patient 13, 1–6 (2020). https://doi.org/10.1007/s40271-019-00399-2. Journal.
- Rhodes LE. Vitamin D status in patients with erythropoietic protoporphyria taking the systemic photoprotective agent afamelanotide. Br J Dermatol. 2024;191(3):317-318. doi:10.1093/bjd/ljae191. PubMed
On DermNet
- Erythropoeitic protoporphyria – pathology
- Photosensitivity
- Porphyria cutanea tarda
- Variegate porphyria
- Acute hepatic porphyrias
- Pseudoporphyia
- Congenital erythropoietic porphyria. Gunther disease.
Other websites
- Cutaneous porphyria — European Dermatology Forum Photosensitivity Guidelines
- Porphyria — Medline Plus
- Porphyria — US National Library of Medicine Genetics Home Reference
- American Porphyria Foundation
- Canadian Porphyria Foundation
- New Zealand Porphyria Support Group (email)
- Porphyria Association Inc. Australia
- Erythropoietic protoporphyria — Medscape Drugs & Diseases
- Erythropoeitic Protoporphyria — British Association of Dermatologists
- European Porphyria Network
- Position statement - Tinted windows — Cancer Council Australia National Cancer Control Policy.
- Porphyria (Cutaneous) Testing Algorithm — Mayo Clinic Medical Laboratories (PDF)