Introduction Demographics Causes Clinical features Complications Diagnosis Differential diagnoses Treatment Outlook
Hereditary coproporphyria (HCP) is a rare subtype of porphyria characterised by a defect in the coproporphyrinogen oxidase (CPOX) enzyme.
It presents with both cutaneous and systemic manifestations. Its rarity and lack of recognition often leads to a low level of clinical suspicion that can delay diagnosis.
The inheritance pattern of the enzyme defect is autosomal dominant but with low clinical penetrance. The mutation can also occur spontaneously.
HCP is caused by a mutation in the coproporphyrinogen oxidase (CPOX) gene leading to a defect in the CPOX enzyme.
The CPOX enzyme is the sixth enzyme involved in the haem (heme) biosynthesis pathway. Most people with HCP have 50% of normal CPOX enzyme activity, although rare homozygous cases with lower levels of CPOX activity have been reported. An absence of CPOX activity is thought to be incompatible with life.
A porphyria attack occurs when there is an upregulation in haem production. This can be triggered by medications, alcohol, cigarette smoking, acute illness, or hormonal changes (eg, pregnancy). The reduced CPOX enzyme level in the affected individual results in accumulation of its substrates – coproporphyrinogen. Coproporphyrinogen then auto-oxidises to coproporphyrin, which is photosensitising and causes cutaneous blistering.
Medications that are considered unsafe in HCP include (not an exhaustive list):
Acute porphyria attacks can present with a range of nonspecific neurovisceral symptoms:
Chronic skin manifestations are also seen, although less commonly than neurovisceral symptoms. These skin changes are the same as those seen with porphyria cutanea tarda (PCT) or variegate porphyria (VP) — subepidermal blisters in sun-exposed skin (eg, back of the hands, face, and neck) with associated scarring, milia development, and/or changes in pigmentation. Hypertrichosis may develop on the temples, cheeks, and dorsal hands.
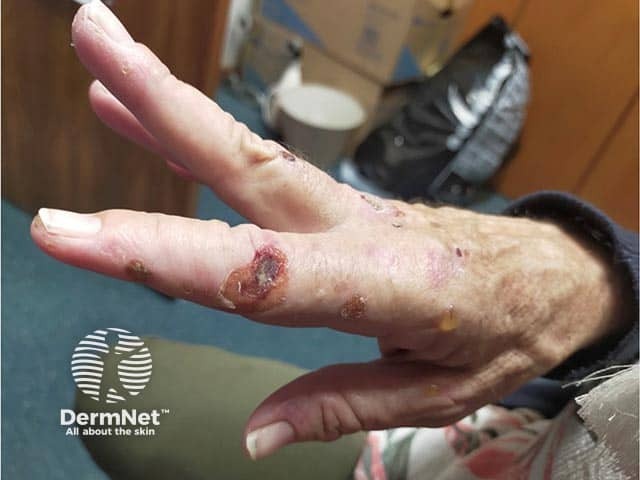
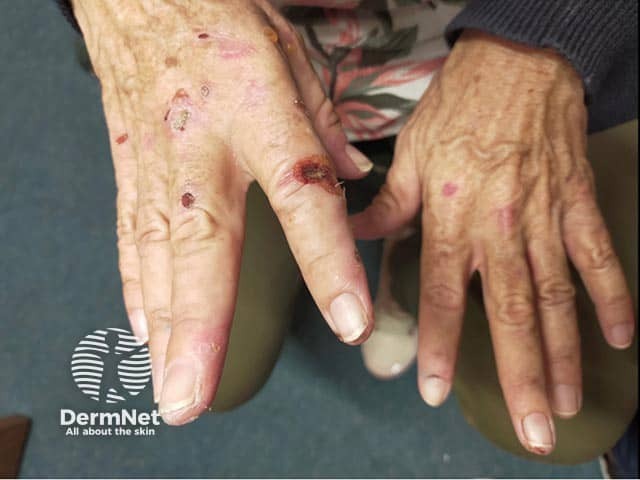
Muscle weakness can lead to flaccid quadriparesis or even respiratory failure, which can be fatal.
HCP can also cause:
People with HCP are at increased risk of hepatocellular carcinoma (HCC), although the exact incidence of this complication cannot be determined due to the rarity of this condition.
Harderoporphyria is a rare form of homozygous HCP complicated by haematological abnormalities.
HCP is diagnosed through biochemical testing of porphyrin levels in the blood (total RBC porphyrins and zinc protoporphyrin), urine (total urinary porphyrins and urinary porphobilinogen), and faeces (total faecal porphyrins).
Other laboratory tests:
Genetic testing looking for the known pathogenic variant of the COPX gene can also confirm the diagnosis. However, it is important to note that a negative genetic test does not exclude the diagnosis.
It is also important to consider and assess for potential triggers (eg, infection, medication) in patients presenting with an acute attack.
Monitoring of urinary and blood porphyrins at least yearly is recommended.
See Acute hepatic porphyrias for more information.
Acute attacks can be life-threatening, although outcomes are improved with early diagnosis and treatment. Rarely, chronic neurological symptoms can develop.
Alcohol abstinence and smoking cessation is advised, along with keeping up-to-date with vaccinations to reduce the risk of infection. Avoidance of triggers is also key in preventing acute porphyria episodes.

