Introduction
Demographics
Causes
Clinical features
Complications
Diagnosis
Differential diagnoses
Treatment
Outlook
Cronkhite–Canada syndrome is a rare, sporadic disorder characterised by generalised gastrointestinal polyposis and dermatological manifestations consisting of hyperpigmentation, hair loss, and nail dystrophy.
Cronkhite–Canada syndrome was first described by Leonard Cronkhite and Wilma Canada in 1955.
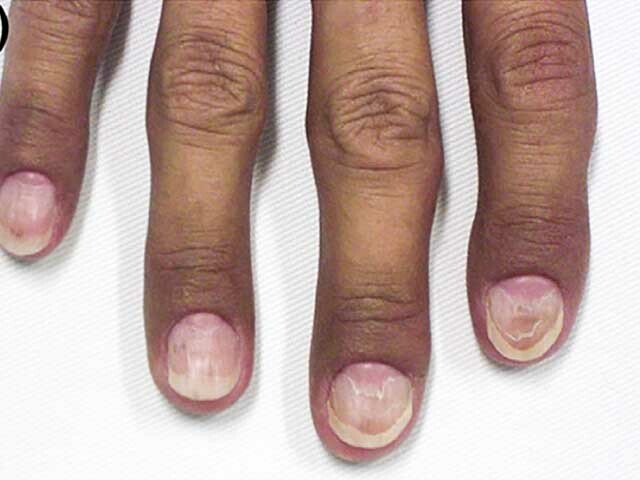
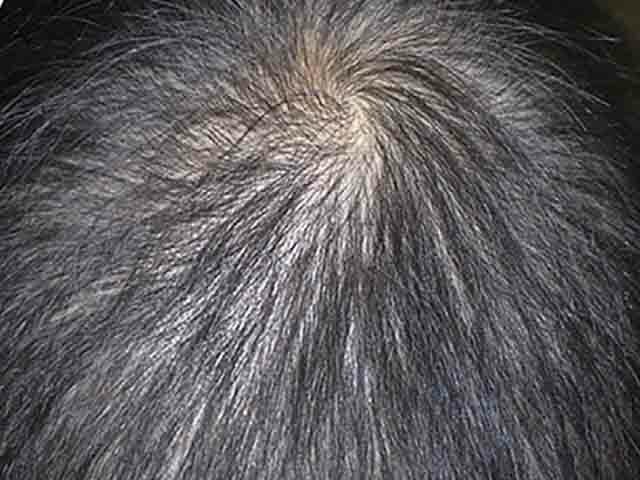
Images from: Onozato Y, Sasaki Y, Abe Y, et al. Cronkhite-Canada syndrome associated with gastric outlet obstruction and membranous nephropathy: a case report and review of the literature. Intern Med. 2020;59(22):2871-7.
The cause of Cronkhite–Canada syndrome is unknown. Physical and emotional stress are commonly reported triggers.
Autoimmune diseases reported in association with Cronkhite-Canada syndrome include vitiligo, Hashimoto thyroiditis, systemic lupus erythematosus, rheumatoid arthritis, and systemic sclerosis. The ANA is often positive.
The cutaneous manifestations may be due to nutritional deficiencies caused by the gastrointestinal changes.
Common presenting symptoms in one large case series were:
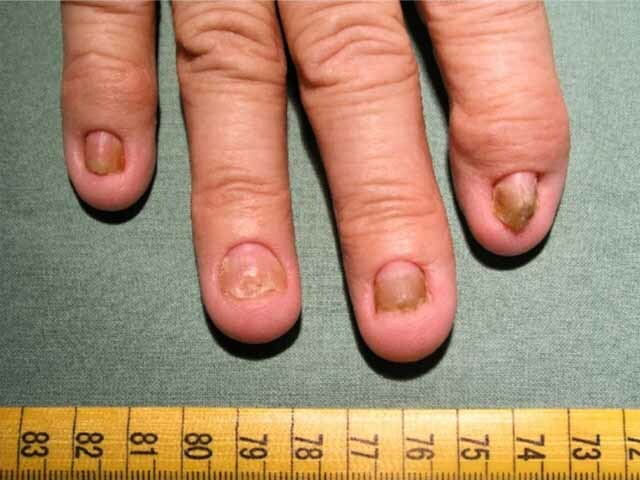
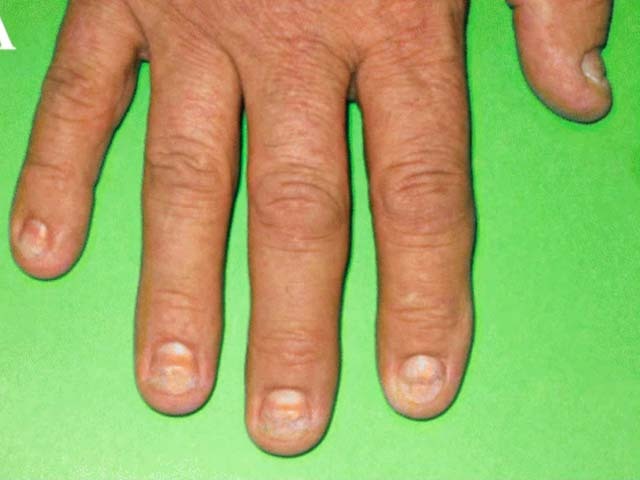
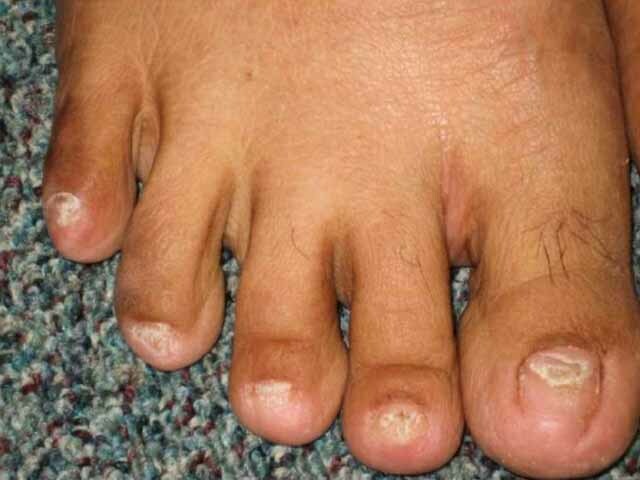
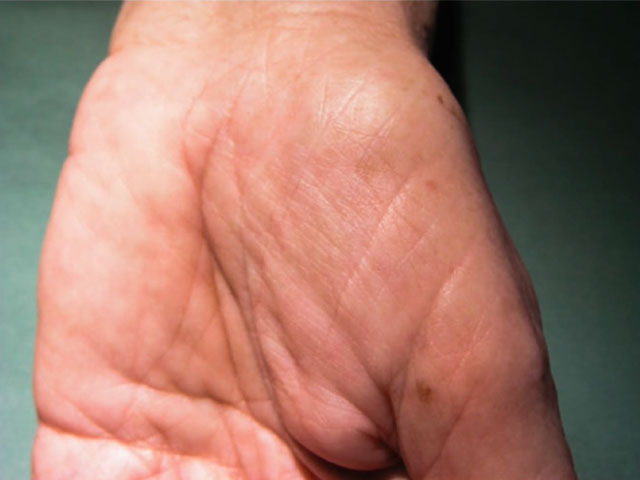
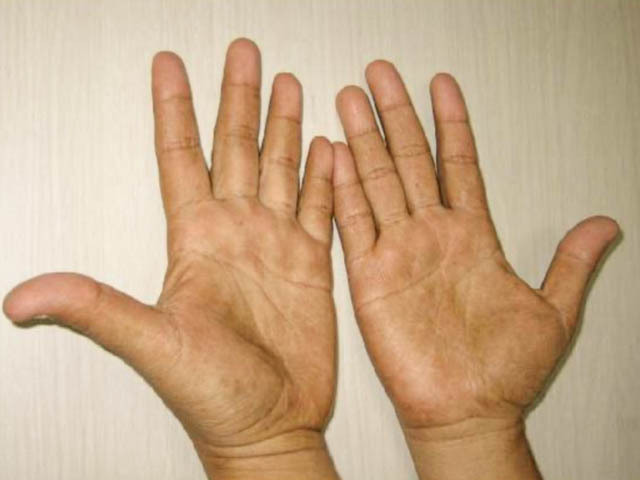
Figures 1&4 from: Kopáčová M, Urban O, Cyrany J, et al. Cronkhite-Canada syndrome: review of the literature. Gastroenterol Res Pract. 2013;2013:856873.
Figure 2 from: Schulte S, Kütting F, Mertens J, et al. Case report of patient with a Cronkhite-Canada syndrome: sustained remission after treatment with corticosteroids and mesalazine. BMC Gastroenterol. 2019;19(1):36.
Figures 3&5 from: Kao KT, Patel JK, Pampati V. Cronkhite-Canada syndrome: a case report and review of literature. Gastroenterol Res Pract. 2009;2009:619378.
The formation of polyps throughout the gastrointestinal tract (but sparing the oesophagus) is the diagnostic finding on endoscopy.
Cronkhite-Canada syndrome should be considered in a patient presenting with the typical gastrointestinal symptoms and skin signs, and confirmed on endoscopy with characteristic histology of dilated mucosal glands, oedematous stroma, and inflammatory infiltrate.
Blood tests will often show anaemia, hypoproteinamia, electrolyte disturbances, nutritional deficiencies, and positive ANA.
Faecal occult blood is positive.
The outlook for Cronkhite–Canada syndrome was initially poor, with mortality rates in the first 5 years after diagnosis of around 50%. Early aggressive treatment with systemic steroids has improved survival and results in the reversal of symptoms and signs in 2-4 months. Steroid dose should be reduced slowly as rebound flare is common if dose reduction is too rapid.

