Introduction Demographics Causes Clinical features Variation in skin types Complications Diagnosis Differential diagnoses Treatment Outcome
Cowden syndrome is a rare, autosomal dominant, inherited condition characterised by hamartomas in various organs, including breast, thyroid, uterus, brain, and mucocutaneous tissues with increased risk of malignancies. It is also known as ‘Cowden disease’ or ‘multiple hamartoma syndrome’.
Cowden disease is one of a spectrum of disorders labelled the PTEN Hamartoma Tumour Syndromes (PHTS), which also include Bannayan-Riley-Ruvalcaba syndrome, Proteus syndrome, and Proteus-like syndrome.
The incidence of Cowden syndrome is 1/200,000, with a female predominance. Most patients have been Caucasian. Skin involvement typically begins in the second or third decade of life.
Cowden syndrome involves the loss of function mutations in the phosphatase and tensin homolog (PTEN) tumour suppressor gene on chromosome 10q23. This results in over-proliferation of cells that form hamartomatous growths.
Approximately 45% of cases may be caused by de novo mutations in PTEN.
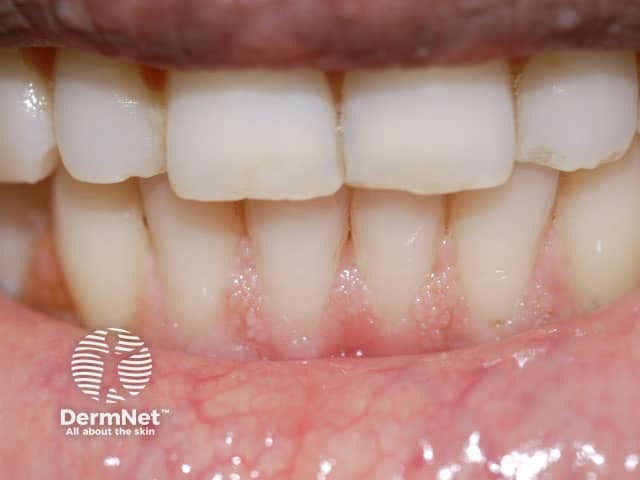
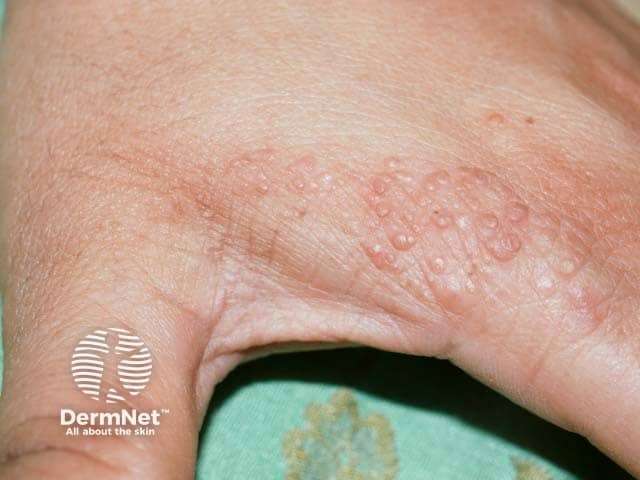
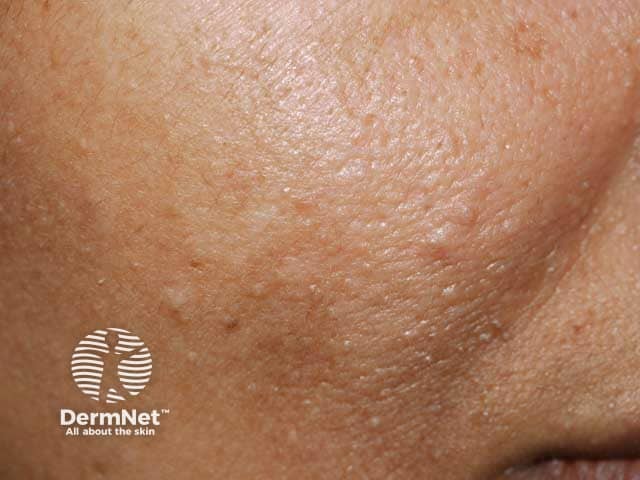
Hamartomas of the skin and mucosa are present in nearly all cases. They may begin in childhood but may not be clearly evident until the second or third decade of life.
Sclerotic fibromas
Oral papillomata
Soft tissue lipomas.
Mucocutaneous neuromas.
Multiple lentigines of the glans penis.
Vascular anomalies such as angiolipomas seen in up to 35%.
Melanoma can be seen in up to 5% of patients.
Glycogenic acanthosis in up to 80%.
The majority of patients reported to have Cowden disease have been Caucasian.
At least 40% of patients with Cowden disease have at least one cancer. Complications will vary depending on the organ system affected by the malignancy.
In 1996, the International Cowden Consortium proposed a set of diagnostic criteria which, with some small adjustments, have been used ever since.
Mucocutaneous lesions:
Major criteria
Minor criteria
Cowden syndrome is diagnosed if a patient has any of the following:
Diagnosis in a family in which one other individual has Cowden disease is made when:
Patients with Cowden disease need to undergo annual medical and physical examinations, laboratory tests, and radiographic tests to regularly check for internal malignancies.
Genetic counselling of relatives is very important, especially for females who are at most risk for malignant complications.
Treatment of the cutaneous features includes:
If cancers are detected early and treated appropriately, the cure rate is high and life expectancy may be close to normal.

