Introduction
Demographics
Causes
Subtypes
Features
Differential diagnoses
Diagnosis
Treatment
Prognosis
Epidermal naevus syndromes are a group of rare disorders characterised by the presence of epidermal naevi (overgrowths of the epidermis) alongside abnormalities in other organs, most often the brain, eyes, and musculoskeletal system.
They are also called systematised epidermal naevi.
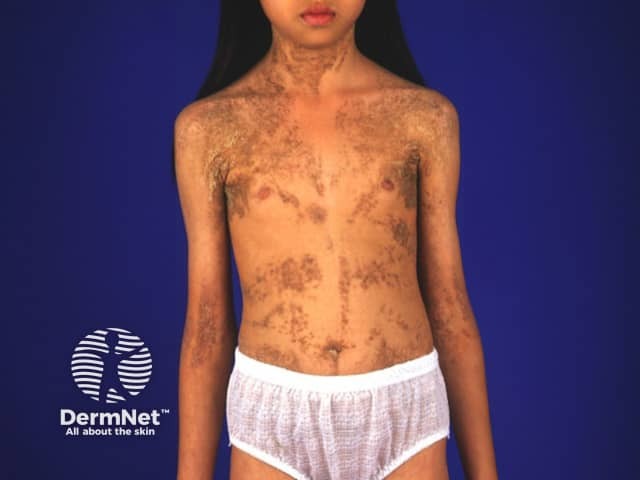
Most available data comes from individual case reports or small case series, making it difficult to determine the epidemiology and true incidence of ENS. However, congenital hemidysplasia with ichthyosiform naevus and limb defect (CHILD) syndrome only affects females due to its X-linked dominant inheritance and is typically male-lethal.
ENS usually arise sporadically due to postzygotic mutations, leading to genetic mosaicism; with the exception of CHILD syndrome which is familial.
As the underlying genetic mechanisms are not fully understood in all cases, classification is often based on the skin pattern and systemic involvement.
Several authors have commented that the term 'epidermal naevus syndrome' is outdated; the classification may change or broaden as more is elucidated about the genetic basis of these conditions.
Several subtypes of ENS have been described, each defined phenotypically by a characteristic type of epidermal naevus and associated systemic features.
Epidermal naevi are categorised as either keratinocytic (linear) or organoid; ENS are similarly categorised. Commonly recognised subtypes include:
Keratinocytic ENS:
Organoid ENS:
Further subtypes eg, Angora hair naevus syndrome, didymosis aplasticosebacea, SCALP syndrome, and others, have been described in isolated case reports. These rarer forms often overlap with the better-defined syndromes or have incomplete genetic characterisation.
CHILD syndrome
CHILD syndrome is an inherited X-linked dominant disorder that almost exclusively affects females. It is caused by mutations in the NSDHL gene, which is involved in the metabolism of cholesterol. The cutaneous and systemic features of CHILD syndrome are typically confined to one side (unilateral) of the body; more often the right hand side.
Cutaneous features:
Systemic features:
Naevus histopathological features: foamy histiocytes in the dermal papillae.
Type 2 segmental Cowden disease (SOLAMEN syndrome)
When Cowden syndrome is associated with a Cowden nevus, this is termed type 2 segmental Cowden disease. This is a mosaic form of Cowden syndrome associated with germline and post-zygotic mutations of the PTEN gene (involved in tumor suppression), presenting with localised skin and systemic features.
The condition is also called SOLAMEN (segmental overgrowth, lipomatosis, arteriovenous malformation, epidermal nevus) syndrome. It often resembles but is genetically distinct to Proteus syndrome.
Cutaneous features:
Systemic features:
Fibroblast growth factor receptor 3 (FGFR3) epidermal nevus syndrome
Also known as García-Hafner-Happle syndrome, this syndrome is caused by a mosaic R248C mutation in the FGFR3 gene.
Cutaneous features:
Systemic features:
Naevus histopathological features: hyperkeratosis, papillomatosis, and acanthosis.
Schimmelpenning syndrome (nevus sebaceous syndrome)
This syndrome is characterised by the presence of sebaceous nevi accompanied by abnormalities of the cardiac, ocular, skeletal, and central nervous systems. Mutations of the HRAS and KRAS genes can cause this syndrome.
Other names include: Schimmelpenning-Feuerstein-Mims syndrome, Jadassohn naevus phakomatosis, organoid naevus phakomatosis, and linear sebaceous naevus syndrome.
Cutaneous features:
Systemic features:
Naevus histopathological features:
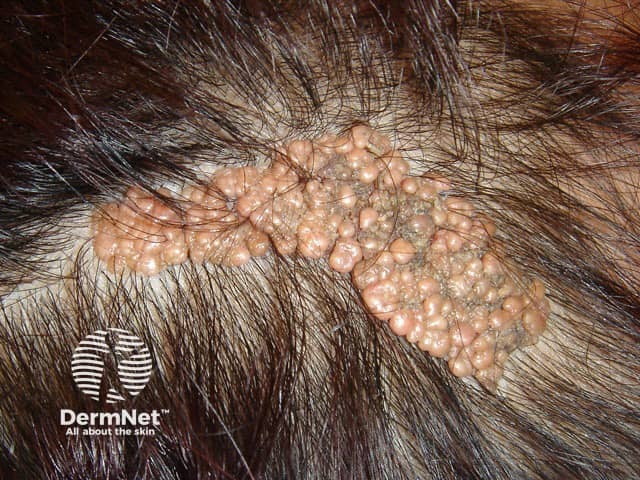
Phakomatosis (sometimes spelt phacomatosis) pigmentokeratotica is a rare ENS representing a form of didymosis (twin spotting), where two genetically distinct naevus types coexist. This syndrome is due to a RASopathy.
Cutaneous features:
Systemic features:
Naevus histopathological features: hyperkeratosis, papillomatosis, and acanthosis.
This refers to the coexistence of a sebaceous naevus with aplasia cutis congenita, typically appearing adjacent to one another. It is thought to result from twin spotting, a form of genetic mosaicism.
SCALP syndrome is didymosis aplasticosebacea (above) in association with a giant melanocytic naevus. The acronym stands for sebaceous naevus, central nervous system abnormalities, aplasia cutis, limbal dermoid, and pigmented naevus.
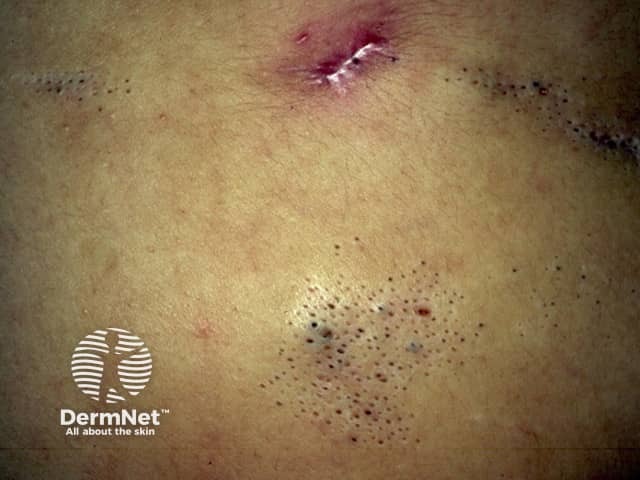
This refers to naevus comedonicus (also known as comedo naevus), presenting with variable extracutaneous abnormalities. Fibroblast growth factor receptor-2 (FGFR2) mutations have been implicated in this syndrome.
Cutaneous features:
Systemic features:
Naevus histopathological features:
This syndrome is characterised by the presence of angora hair naevi, soft, long-haired lesions resembling Angora wool.
Cutaneous features:
Systemic features:
Naevus histopathological features: hyperpigmentation of the basal layer and acanthosis.
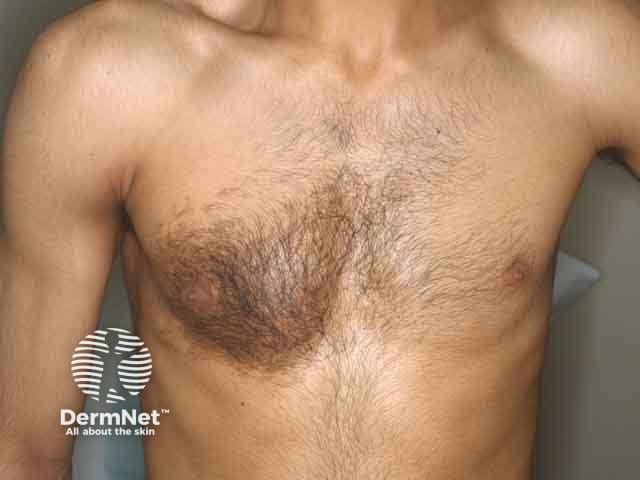
This syndrome is characterised by the presence of a Becker naevus (a pigmented, hairy patch) and other systemic abnormalities.
Cutaneous features:
Systemic features:
Naevus histopathological features:
Several dermatological and syndromic conditions can resemble epidermal naevus syndromes and should be considered in the differential diagnosis. These include:
Diagnosis is primarily clinical, based on a detailed history and physical examination. In patients with systemic involvement, supportive investigations may include:
There is no cure. A multidisciplinary approach is often necessary to optimise the management of symptoms.
Prognosis depends on the severity and extent of extracutaneous involvement. Individuals with isolated epidermal naevi generally have a good prognosis, with most concerns being cosmetic. However, syndromic cases with neurological or organ involvement may face more complex challenges.

