Introduction Demographics Causes Clinical features Variations Diagnosis Histopathological features Differential diagnoses Complications Prevention Outlook
A hamartoma is a benign (non-cancerous) overgrowth of a mature cell type normal to the site, tissue, or organ. The word is from the Greek hamartia (‘fault’ or ‘defect’) and -oma (‘tumour’ or ‘neoplasm’).
In the skin, a hamartoma often presents as a birthmark (congenital naevus), but may develop in adulthood (acquired).
In general, hamartomas affect those aged 40–70, and males more than females. The incidence of many hamartomas is unknown and varies depending on type. The reported incidence rate of pulmonary hamartoma is 0.25%, while that of breast hamartoma is 0.1–0.7%.
Hamartomas are more common in conditions such as tuberous sclerosis complex and Cowden syndrome. Other risk factors include age, hormonal factors, obesity, family history, and genetic predisposition.
Some hamartomas are more commonly encountered in children. These include cardiac hamartomas, lipofibromatous hamartoma of the nerve, hypothalamic hamartoma, and Peutz-Jeghers syndrome.
Unlike a malignant tumour, hamartomas usually originate from the overgrowth of multiple aberrant cells rather than a singular mutated cell. Hamartomas cause a structurally disorganised malformation local to the original tissue but are non-neoplastic and rarely invade or significantly compress surrounding structures. It is possible to have neoplastic evolution within some hamartomas.
The underlying mechanisms of hamartomas are not fully understood. They are present in a broad spectrum of disorders and syndromes. Certain gene mutations have been associated with their development, including PTEN, SMAD4, STK1, BMPR1A, TSC-1, and TSC-2.
Hamartomas are distinctly demarcated masses that contain a variety of cell types depending upon their origin. Size is variable, mostly ranging between 1–4 cm, while some pulmonary hamartomas can be as large as 10 cm.
Cutaneous hamartomas may be verrucous, sebaceous, fibrous, achromatic, pilar, or other.
Clinical features vary depending on the location of the hamartoma and, consequently, the mass effect or localised pathology that may arise.
Other potential symptoms include abdominal or flank pain, fevers, night sweats, and breast or testicular enlargement.
Below are examples of different types of hamartomas.
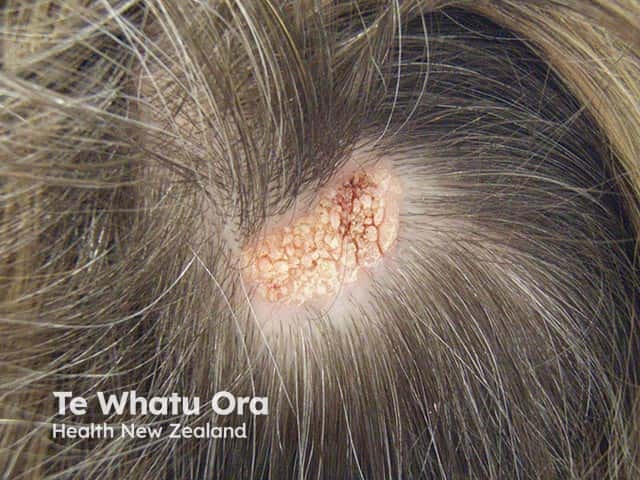
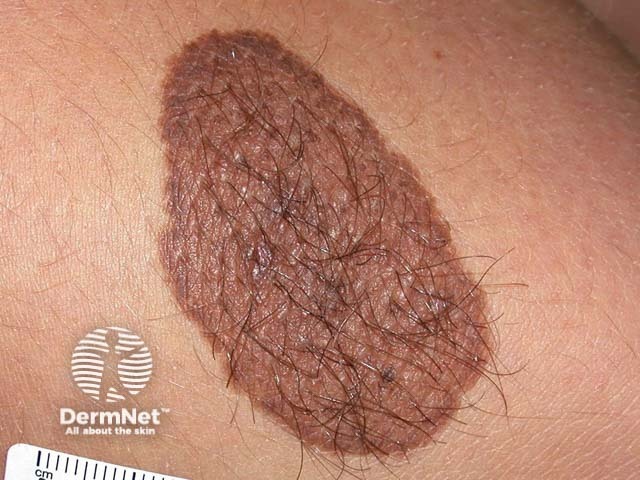
Diagnosis of a hamartoma varies depending on the type. In general, they are often diagnosed with a combination of laboratory testing, imaging (eg, x-ray, ultrasound, CT scan, or MRI), and biopsy for tissue diagnosis.
On histopathological examination, hamartomas resemble benign tumours, and may include disorganised overgrowth of normal tissue. The cells cytologically resemble local tissues, and may be well-circumscribed.
There are not usually any signs of invasion into surrounding tissue or metastasis.
Specific histopathological characteristics depend on the type of hamartoma, for example:
Differential diagnoses of a hamartoma include different types of benign or malignant tumours. The types of growths or neoplasia considered may be specific to the site involved.
Hamartomas are usually asymptomatic. However, some can grow very large leading to disfigurement and pressure on surrounding structures. Depending on location, this can lead to complications such as seizures, airway obstruction, abdominal obstruction, heart failure, or breast deformity. Some hamartomas can also lead to infection, infarction, fracture, haemorrhage, or (rarely) neoplastic transformation.
It is not known how hamartomas can be prevented.
Hamartomas are almost always benign and many are asymptomatic, although rarely malignant transformation may occur. Prognosis varies depending on the size and location of the hamartoma, as well as the health status and comorbidities of the patient.

