

Vogt–Koyanagi–Harada syndrome — extra information
Introduction
Demographics
Causes
Clinical features
What are the dermatological findings in Vogt-Koyanagi-Harada syndrome?
Complications
Diagnosis
Differential diagnoses
Treatment
Outcome
What is Vogt–Koyanagi–Harada syndrome?
Vogt-Koyanagi-Harada syndrome (VKH) is a multisystem disease that presents with a combination of ophthalmological, neurological, and dermatological signs and symptoms.
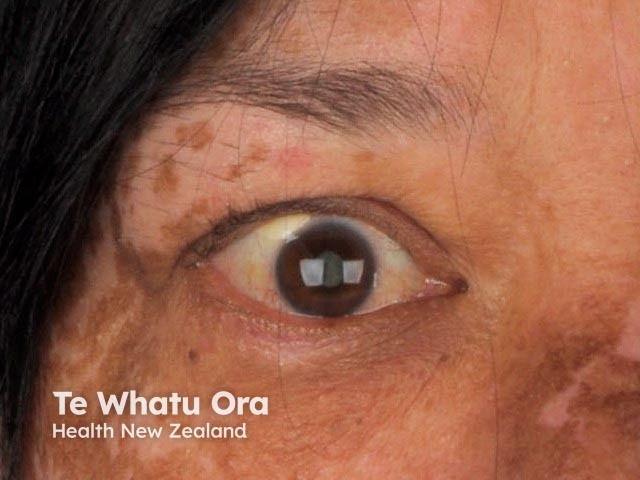
- The main defining clinical finding of VKH is severe bilateral granulomatous panuveitis (inflammation throughout the uveal tract in the eye).
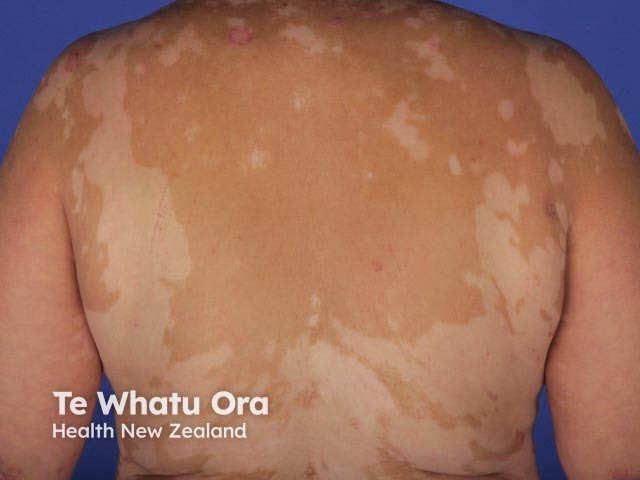
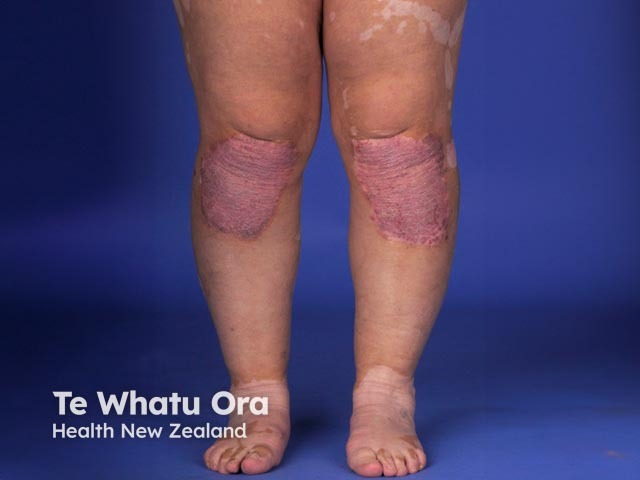
- Cutaneous features include vitiligo, alopecia areata, and poliosis (white hair).
- Neurological features include meningism (symptoms of meningitis with no inflammation), hearing loss, and tinnitus.
Who gets Vogt–Koyanagi–Harada syndrome?
VKH is reported to have significant regional and global variation [1]. VKH is more prevalent in patients with a darker skin pigmentation than in Caucasians and people of Turkish descent. Patients with VKH are most commonly of Asian, Hispanic, Indian, Native American, or Mediterranean origin [2]. There are twice as many females affected by VKH as males [3].
VKH is mainly diagnosed in adult and middle-aged patients aged 20–50, although onset in childhood or old age has also been reported [4–6].
What causes Vogt–Koyanagi–Harada syndrome?
The exact cause of VKH is yet to be established. It is an autoimmune inflammatory condition mediated by CD4+ T cells targeting melanocytes [7,8]. Genetic predisposition and/or an unspecified exposure to a virus may be involved [1].
What are the clinical features of Vogt-Koyanagi-Harada syndrome?
Classically, VKH has four clinical stages. The clinical features vary, depending on ethnic group. Dermatological features are most often noted in the chronic convalescent stage [1].
The prodromal stage
The prodromal stage of VKH may mimic viral infection and lasts for 1–2 weeks [2]. The symptoms are mainly neurological, and include:
- Headache
- Neck stiffness
- Photophobia
- Fever
- Orbital pain
- Scalp and general skin sensitivity
- Focal neurological deficits (eg, cranial nerve palsy).
Auditory symptoms include:
- Hearing loss (particularly for higher frequencies)
- Discomfort when exposed to sounds at normal volume
- Vertigo
- Tinnitus [2].
The acute stage
The acute stage of VKH affects the eye and lasts for several weeks [2].
- The main symptom of the acute stage of VKH is blurred vision in both eyes [1].
- Ophthalmological findings are panuveitis or posterior uveitis, with multifocal serous retinal detachments [2].
- Early recognition and treatment can prevent progression to chronic convalescent and chronic recurrent stages of the disease [7].
The chronic convalescent stage
Many patients with acute VKH go on to the chronic convalescent stage of VKH, about 3–4 months after the onset of the disease [9,10]. This usually lasts for some months or years [1,2].
- Ophthalmological findings of the chronic convalescent phase of VKH include non-granulomatous panuveitis and depigmentation of the choroid (the middle coat of the eye), with a bright orange fundus (a 'sunset glow') [10,11].
- The dermatological findings are poliosis of the eyebrows and eyelashes, vitiligo, and alopecia areata [1].
The chronic recurrent stage
25% of patients with acute VKH develop the chronic recurrent phase of the disease 6–9 months after initial presentation [1,2].
- Ophthalmological findings are recurrent granulomatous anterior uveitis and choroidal thickening.
- Vitiligo and alopecia areata may have phases of relapse and remission.
What are the dermatological findings in Vogt–Koyanagi–Harada syndrome?
About 30% of patients with VKH develop dermatological findings [12]. Vitiligo, poliosis, and alopecia areata usually occur in the chronic convalescent stage of the disease [13].
What are the complications of Vogt–Koyanagi–Harada syndrome?
Patients with VKH can develop complications that threaten or impair vision, such as:
- Retinal detachment in the acute phase [2]
- Secondary glaucoma in the chronic phase (45% of VKH cases)
- Cataracts (42% of cases [11,14,15])
- Subretinal fibrosis (40% of cases [16]).
How is Vogt-Koyanagi-Harada syndrome diagnosed?
The diagnostic criteria for VKH include inflammation of both eyes without evidence of another ocular disease, a history of trauma, or ocular surgery [17].
There are three distinct categories of the disease [17]:
- Complete VKH — where there is bilateral ocular involvement (with detailed criteria depending on the stage of disease), plus neurological and dermatological signs; neurological signs may resolve, whereas dermatological signs do not precede the onset of uveitis.
- Incomplete VKH — where the ophthalmological findings are as for patients with complete VKH, plus either neurological or dermatological signs.
- Probable VKH — where the ophthalmological findings are as for patients with complete VKH, but without neurological or dermatological signs.
What is the differential diagnosis for Vogt–Koyanagi–Harada syndrome?
Differential diagnoses for the ophthalmological signs and symptoms of the disease include:
- Prior trauma — including sympathetic ophthalmia (inflammation of the conjunctiva or the eyeball; sympathetic ophthalmia can have an almost identical clinical picture to VKH, including its dermatological manifestations [18]
- Infection — including bacterial or fungal infection, tuberculosis or syphilis [1]
- Malignancy — such as intraocular lymphoma, systemic lymphoma, or leukaemia [1]
- Inflammatory disease — including bilateral posterior scleritis, sarcoidosis, or lupus choroidopathy (degradation of the eye coating due to lupus) [1].
What is the treatment for Vogt–Koyanagi–Harada syndrome?
Acute VKH should be treated aggressively with a systemic corticosteroid (minimum treatment duration is 6 months), combined with a corticosteroid-sparing agent, such as ciclosporin, methotrexate, or mycophenolate mofetil [1]. Anti–tumour necrosis factor alpha (TNF) agents or other biological agents can be considered in patients who do not respond to corticosteroids combined with corticosteroid-sparing agents [1].
The aim of treatment is to dampen the inflammatory response and to prevent ocular complications, such as sunset glow fundus, cataracts, glaucoma, and choroidal neovascularisation [1].
What is the outcome for Vogt–Koyanagi–Harada syndrome?
Better visual outcomes are described in those patients who undergo prompt and aggressive treatment of VKH. However, a significant proportion of patients still develop vision-impairing complications. Cataracts and glaucoma often require surgical management [1,2].
Favourable prognostic factors include good initial visual acuity and initiation of treatment in the acute rather than chronic recurrent stage of the disease [11,15,19].
References
- O'Keefe G, Rao N. Vogt-Koyanagi-Harada disease. Surv Ophthalmology 2017; 62: 1–25. DOI: doi.org/10.1016/j.survophthal.2016.05.002. Journal
- Baltmr A, Lightman S, Tomkins-Netzer O. Vogt–Koyanagi–Harada syndrome — current perspectives. Clin Ophthalmol 2016; 10: 2345–61. DOI: 10.2147/OPTH.S94866. PubMed
- Wang Y, Chan C. Gender Differences in Vogt–Koyanagi–Harada disease and sympathetic ophthalmia. J Ophthalmol 2014; 2014: Article ID 157803 [8 pages]. DOI: dx.doi.org/10.1155/2014/157803. Journal
- Rao N, Moorthy R, Inomata H. Vogt–Koyanagi–Harada syndrome. Int Ophthalmol Clin 1995; 35: 69–86. Journal
- Abu El-Asrar A, Al-Kharashi A, Aldibhi H, Al-Fraykh H, Kangave D. Vogt–Koyanagi–Harada disease in children. Eye 2008: 22: 1124–31. DOI:10.1038/sj.eye.6702859. Journal
- Yamamoto Y, Fukushima A, Nishino K, Koura Y, Komatsu T, Ueno H. Vogt-Koyanagi-Harada Disease with onset in elderly patients aged 68 to 89 years. Jpn J Ophthalmol 2007; 51: 60–3. DOI: doi.org/10.1007/s10384-006-0379-0. Journal
- Norose K, Yano A. Melanoma specific Th1 cytotoxic T lymphocyte lines in Vogt-Koyanagi-Harada disease. Br J Ophthalmol 1996; 80: 1002–8. DOI: dx.doi.org/10.1136/bjo.80.11.1002. Journal
- Sugita S, Sagawa K, Mochizuki M, Shichijo S, Itoh K. Melanocyte lysis by cytotoxic T lymphocytes recognizing the MART-1 melanoma antigen in HLA-A2 patients with Vogt–Koyanagi–Harada disease. Int Immunol 1996: 8: 799–803. DOI: doi.org/10.1093/intimm/8.5.799. Journal
- Chee S, Jap A, Bacsal K. Spectrum of Vogt-Koyanagi-Harada disease in Singapore. Int Ophthalmol 2006: 27: 137–42. Journal
- Keino H, Goto H, Usui M. Sunset glow fundus in Vogt-Koyanagi-Harada disease with or without chronic ocular inflammation. Graefes Arch Clin Exp Ophthalmol 2002; 240: 878–82. DOI: 10.1007/s00417-002-0538-z. Journal
- Read R, Rechodouni A, Butani N, et al. Complications and prognostic factors in Vogt–Koyanagi–Harada disease. Am J Ophthalmol 2001: 131: 599–606. PubMed
- Yang P, Ren Y, Li B, Fang W, Meng Q, Kijlstra A. Clinical characteristics of Vogt–Koyanagi–Harada syndrome in Chinese patients. Ophthalmology 2007: 114: 606–14. DOI: 10.1016/j.ophtha.2006.07.040. Journal
- Beniz J, Forster D, Lean J, Smith R, Rao N. Variations in clinical features of the Vogt-Koyanagi-Harada syndrome. Retina 1991: 11: 275–80. Europe PMC
- Rubsamen PE, Gass JDM. Vogt–Koyanagi–Harada syndrome. Clinical course, therapy, and longterm visual outcome. Arch Ophthalmol 1991: 109: 682–7. DOI:10.1001/archopht.1991.01080050096037. Journal
- Arevalo J, Lasave A, Gupta V, et al. Clinical outcomes of patients with Vogt–Koyanagi–Harada disease over 12 years at a tertiary center. Ocul Immunol Inflamm 2015; 24: 521–9. DOI: 10.3109/09273948.2015.1025984. Journal
- Lertsumitkul S, Whitcup SM, Nussenblatt RB, Chan CC. Subretinal fibrosis and choroidal neovascularization in Vogt–Koyanagi–Harada syndrome. Graefe's Arch Clin Exp Ophthalmol 1999; 237: 1039–45. Journal
- Read R, Holland G, Rao N, et al. Revised diagnostic criteria for Vogt–Koyanagi–Harada disease: report of an international committee on nomenclature. Am J Ophthalmol 2001: 131: 647–52. DOI:).1016/S0002-9394(01)00925–4. Journal
- Rao N, Gupta A, Dustin L. Sympathetic ophthalmia simulating Vogt-Koyanagi-Harada's disease: a clinico-pathologic study of four cases. Jpn J Ophthalmol 1983; 27: 506–11. Journal
- Abu El-Asar AM, Al Tamimi M, Hemachandran S, Al-Mezaine HS, Al-Muammar A, Kangrave D. Prognostic factors for clinical outcomes in patients with Vogt-Koyanagi-Harada disease treated with high-dose corticosteroids. Acta Ophthalmol 2013; 91: 486–93. DOI: doi.org/10.1111/aos.12127. Journal
On DermNet
Other websites
- Vogt-Koyanagi-Harada disease — Medscape
- Vogt-Koyanagi-Harada disease — National Organization for Rare Disorders (NORD)