Introduction
Demographics
Causes
Clinical features
Complications
Diagnosis
Differential diagnoses
Treatment
Outcome
Amyloidosis cutis dyschromica is a form of primary cutaneous amyloidosis that causes localised hyperpigmentation and hypopigmentation.
Amyloidosis cutis dyschromica was first described by Japanese dermatologist Morishima in 1970 [1].
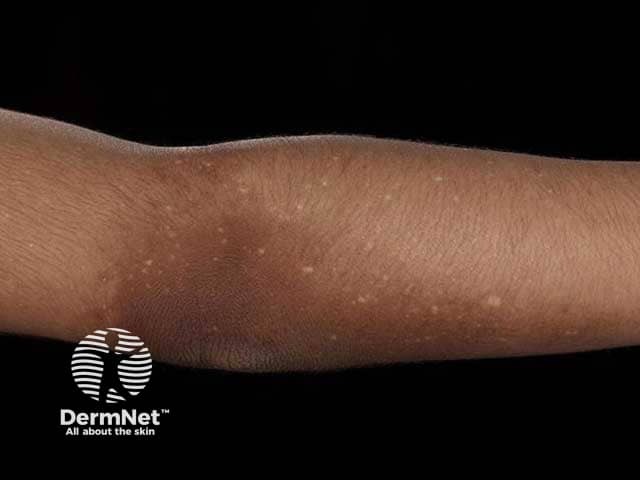
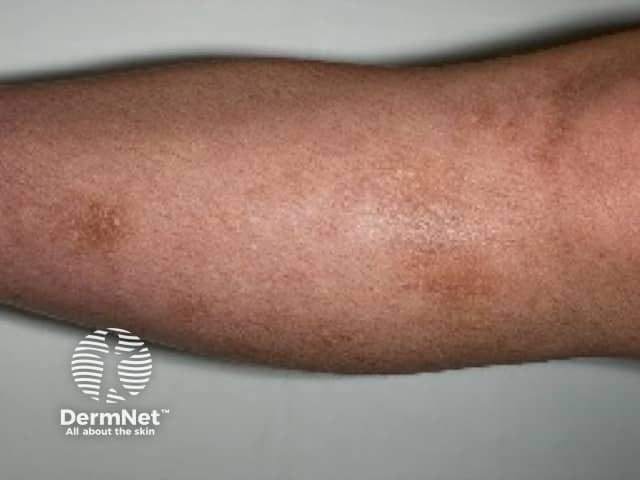
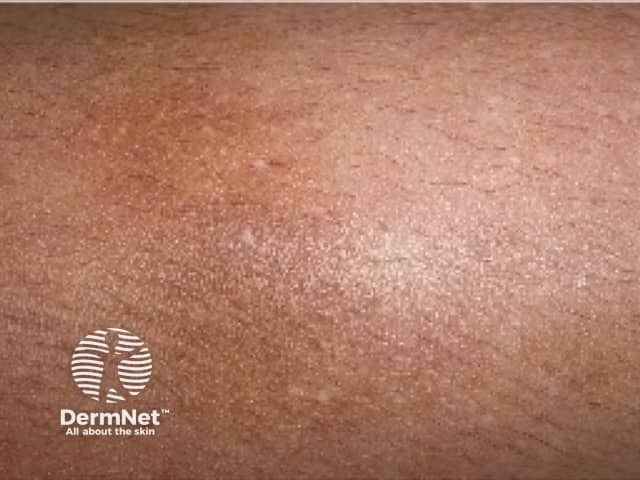
Amyloidosis cutis dyschromica is rare. At the time of writing, about 50 cases have been reported in the medical literature [2].
Amyloidosis cutis dyschromica is caused by the deposition of amyloid derived from degenerate or damaged keratinocytes. The reason why the keratinocytes degenerate and how this relates to the loss and gain of pigment in the affected areas of skin is not clear. A genetic cause is likely.
Amyloidosis cutis dyschromica causes slowly progressive localised hyperpigmentation and hypopigmentation (dyschromica or dyschromatosis).
The specific findings that distinguish amyloidosis cutis dyschromica from the more common macular and lichenoid variants of primary cutaneous amyloidosis are:
There are no known complications from amyloidosis cutis dyschromica.
Amyloidosis cutis dyschromica is diagnosed by recognising the typical clinical features and confirmed by the biopsy of a hyperpigmented or hypopigmented macule.
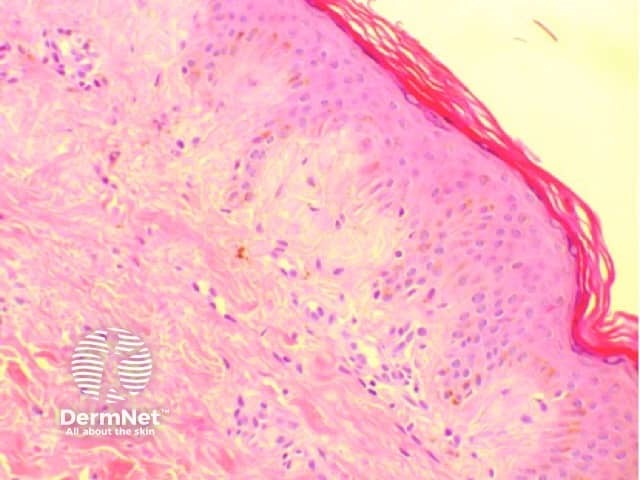
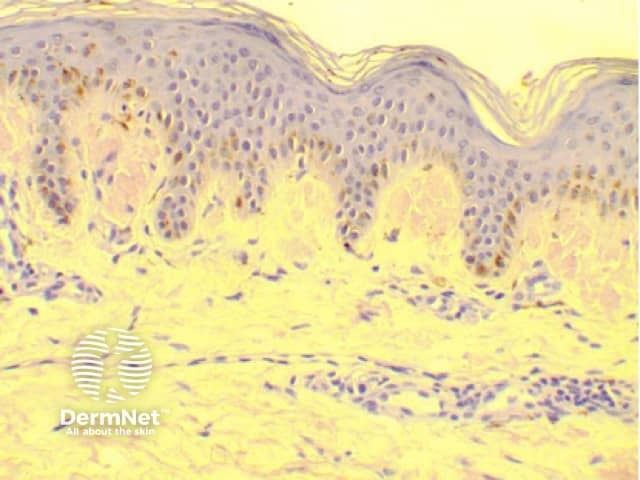
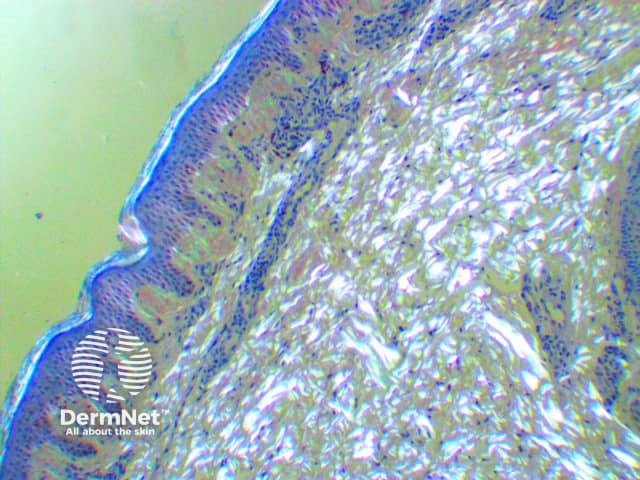
Amyloidosis cutis dyschromica may closely resemble a number of other rare dyschromatoses; these include:
Other conditions that should be excluded include:
No therapeutic intervention has been demonstrated to be of benefit in amyloidosis cutis dyschromica. Multiple topical treatments, including 10% urea cream and tazarotene [1], have been prescribed to patients without significant improvement. Oral vitamin C and vitamin E supplements have had minimal benefit [3]. Acitretin has been prescribed in a small number of cases, with a modest improvement reported in approximately 85% of cases [2].
Amyloidosis cutis dyschromica is typically gradually progressive. The dyschromia may eventually involve almost all of the skin except the palms, soles of the feet, and oral and genital mucosa. Alternatively, amyloidosis cutis dyschromica can progress gradually in localised areas, and it is often more prominent in areas of skin overlying bony prominences and joint surfaces.

