Introduction
Demographics
Clinical features
Diagnosis
Assessment of severity
Treatment
Epidermolysis bullosa (EB) is a group of inherited diseases that are characterised by blistering lesions on the skin and mucous membranes. These may occur anywhere on the body but most commonly appear at sites of friction and minor trauma such as the feet and hands. In some subtypes, blisters may also occur on internal organs, such as the oesophagus, stomach and respiratory tract, without any apparent friction.
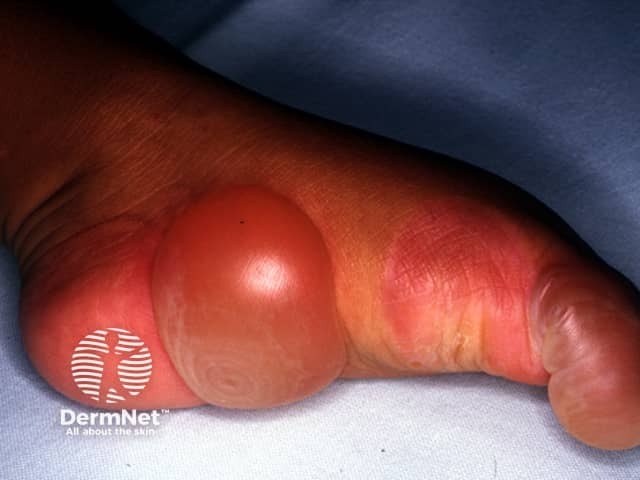
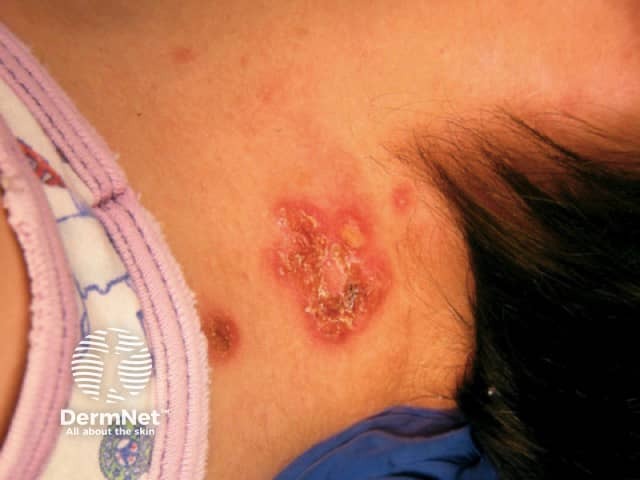
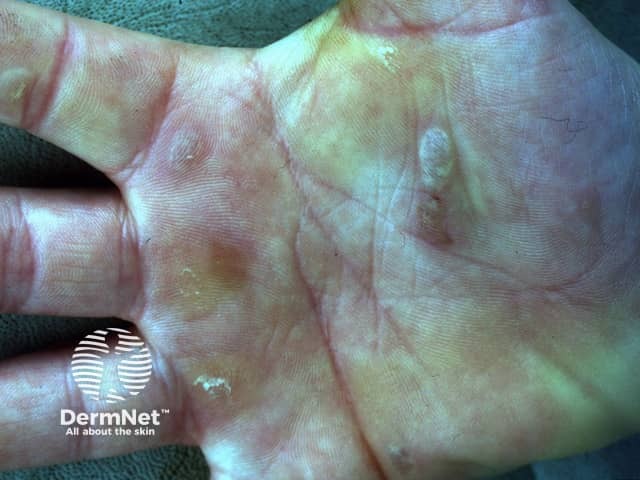
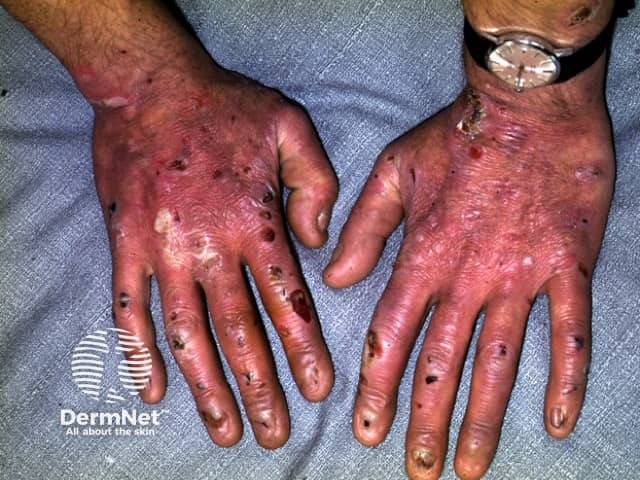
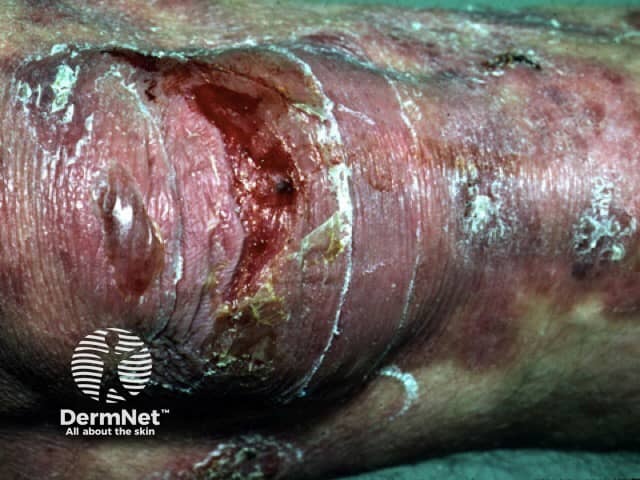
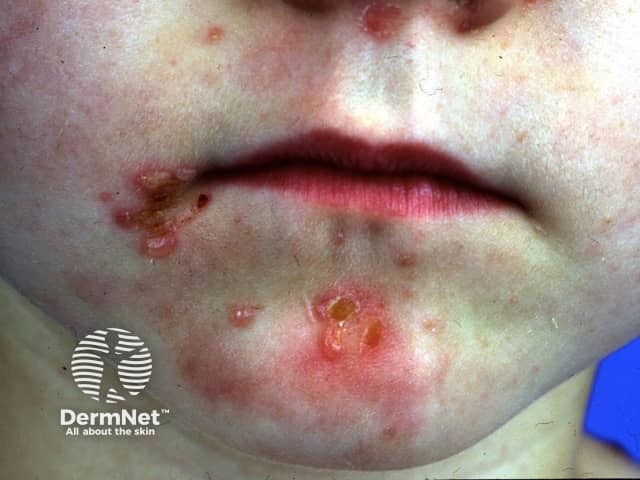
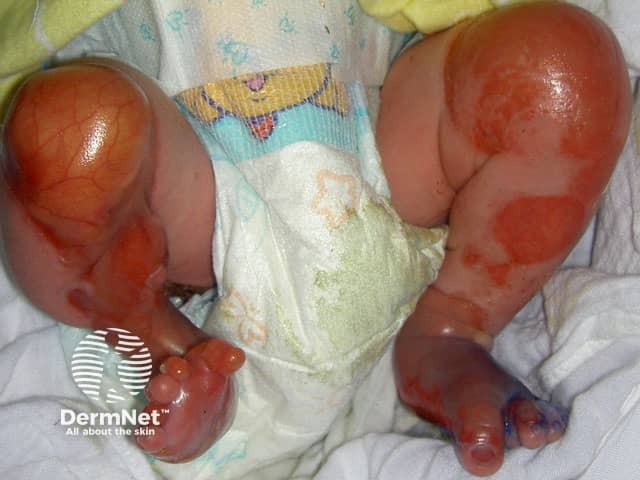
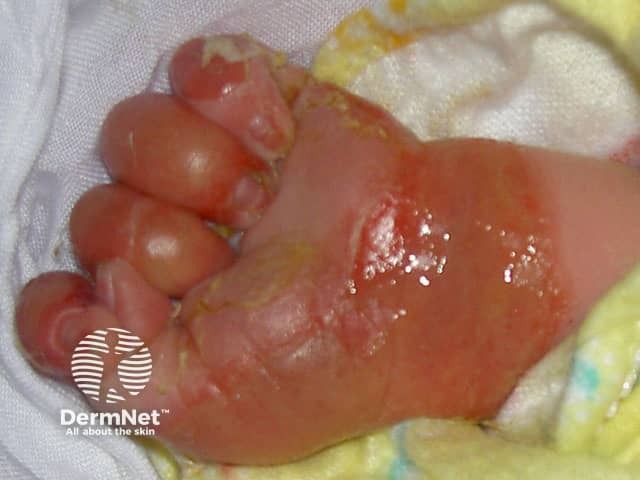
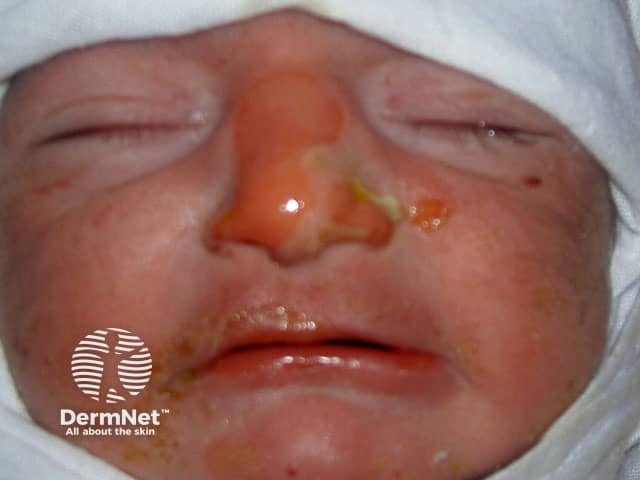
EB should be distinguished from common friction blisters, and from epidermolysis bullosa acquisita (EBA), which is a blistering autoimmune disease that is not inherited and often doesn't develop until adult life.
The EB conditions result from genetic defects of molecules in the skin concerned with adhesion. Loss of adhesion results in blister formation. There are four major types of EB based on different sites of blister formation within the skin structure:
EB type |
The site of blister formation within skin |
|---|---|
Epidermis or uppermost layer of skin cells (keratinocytes) |
|
Lamina lucida within the basement membrane zone |
|
Lamina densa and upper dermis |
|
Mixed pattern or multiple levels within and beneath the basement membrane zone |
Within each of these types of EB, there are various subtypes. Varying degrees of severity that range from mild to severe are found with each EB type. International consensus on the diagnosis and classification of EB has resulted in updated recommendations (2014), based on newer clinical and molecular data.
EB is an inherited disease, which means that you have inherited one or two EB genes. In autosomal dominant EB, only one abnormal gene is needed to express the disease. This means only one parent needs to carry the EB gene. On the other hand, autosomal recessive inherited EB requires you to have two EB genes (one from each parent) to have the disease. If a person has one recessive EB gene paired with a normal gene they are called a carrier and do not have the disease.
EB usually occurs at birth or shortly after. Males and females are equally affected. Occasionally EB may be mild enough at birth not to be apparent, and it is not until the child is older or reaches adulthood before it is detected.
For more details, see Epidermolysis bullosa simplex.
EBS Subtypes |
Features |
|---|---|
Localised EBS |
|
Generalised EBS |
|
Generalised severe EBS |
|
For more details, see Junctional epidermolysis bullosa.
JEB Subtypes |
Features |
|
|---|---|---|
Generalised severe JEB |
|
|
Generalised intermediate JEB |
|
|
For more details, see Dystrophic epidermolysis bullosa.
DEB Subtypes |
Features |
|
|---|---|---|
Dominant generalised DEB |
|
|
Generalised severe recessive (R) DEB |
|
|
Kindler syndrome |
Features |
|
|---|---|---|
Kindler syndrome |
|
|
There are many other subtypes of EB. The presentation and severity of EB are affected by the specific genetic changes and can at times be difficult to classify.
Genetic counselling and prenatal testing is available for EB in some centres and should be considered.
Referral for psychological support should also be considered for children with EB and their family, especially for the more severe subtypes.
In the dominant subtypes of EB, where an informative family tree is known, it is often acceptable for a clinical diagnosis (based on the presenting signs) to be made by a specialist dermatologist.
Diagnostic tests are also available in some countries and include skin biopsy of a newly induced blister which undergoes immunofluorescence antigen mapping (IFM) and transmission electron microscopy (EM). Mutational analysis (blood testing of genes), although not currently considered the first-line diagnostic test, is also available in some countries.
A referral to a dermatologist will be required.
The severity of EB can be assessed using the following scoring systems:
There is no cure for EB. However, significant research, including gene therapy and cell-based therapy, continue in the aim to improve quality of life.
Most current treatment is symptomatic. The primary aim is to protect the skin and stop blister formation, promote healing, and prevent complications.
Because EB can affect so many different parts of the body, a team of medical specialists is usually required for overall care. When necessary, treatment with oral and topical medications may be prescribed by your doctor to assist healing or prevent complications.
The following are some general measures used in caring for a patient with EB.

