Introduction - epidermolysis bullosa
Introduction
Demographics
Causes
Clinical features
Diagnosis
Differential diagnoses
Treatment
Prognosis
Epidermolysis bullosa acquisita (EBA) is a rare, acquired, autoimmune, mucocutaneous blistering disease that occurs at sites of minor trauma.
Unlike epidermolysis bullosa, EBA is not inherited and usually presents in adulthood.
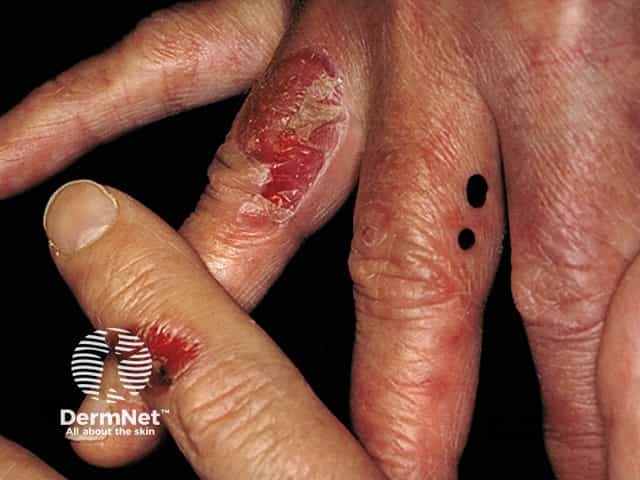
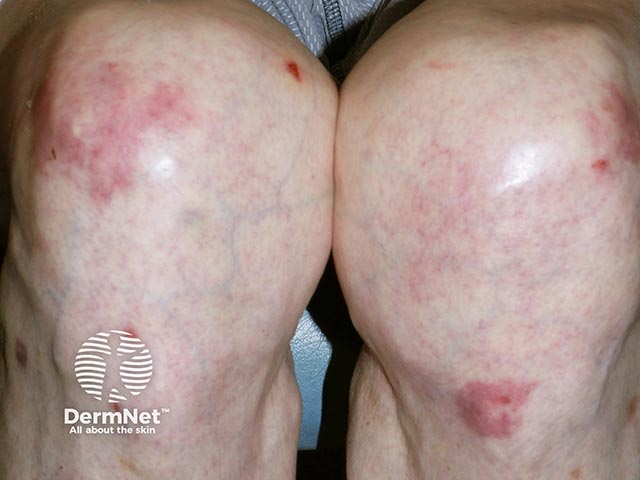
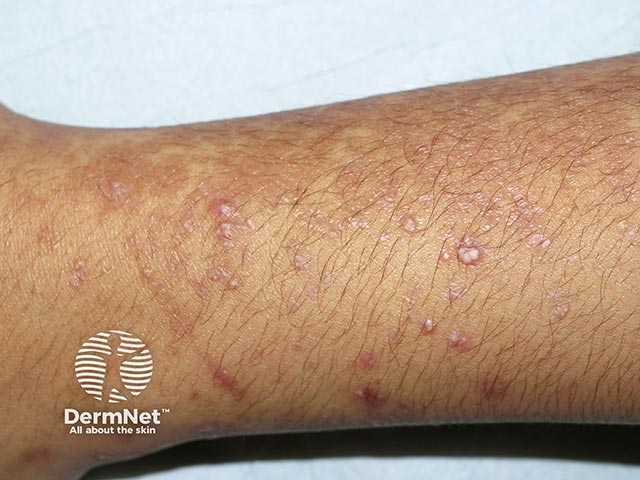
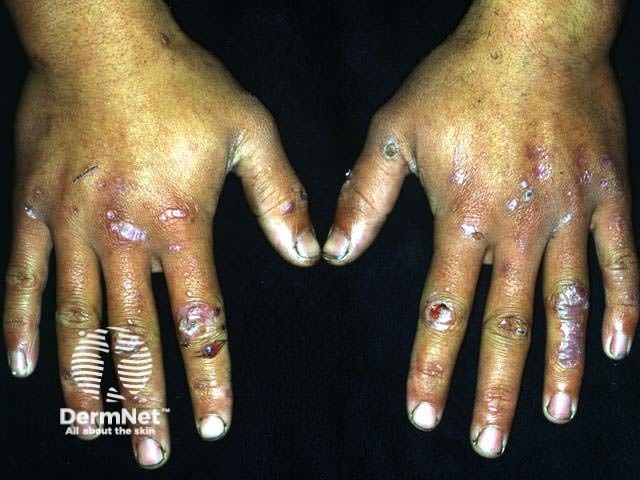
EBA is considered one of the rarest subepidermal blistering diseases. The exact incidence and prevalence remain unknown, though the annual incidence is estimated to be less than 0.5 cases per million individuals. While it typically affects adults (50-70 years old), paediatric cases have been reported, including a neonatal case caused by transfer of antibodies across the placenta.
Gender, racial, ethnic, or geographical predispositions have not been conclusively established. However, some studies report a greater prevalence amongst certain African-American populations.
Patients with EBA may have concomitant systemic disorders such as:
The relationship between EBA and these disorders remains unclear.
While the exact aetiology remains unknown, epidermolysis bullosa acquisita is considered an autoimmune disorder characterised by tissue-bound autoantibodies (IgG) against the NC1 domain of type VII collagen.
Type VII collagen is the major structural component of anchoring fibrils, which attach the basement membrane to the underlying papillary dermis at the dermo-epidermal junction. Loss of fibrils due to autoimmune inflammation and destruction results in subepidermal blistering within the lamina densa.
Potential triggering factors reported in the literature include:
Genetics may also play a role in the development of EBA, as human leukocyte antigen (HLA) class II genotypes are more common amongst those with EBA.
Epidermolysis bullosa acquisita can be categorised into two common presentations: classical (mechanobullous) EBA and inflammatory (non-mechanobullous) EBA.
A mixture of inflammatory and non-inflammatory features may co-exist.
Inflammatory EBA variants |
Clinical features |
|||||
|---|---|---|---|---|---|---|
Bullous pemphigoid-like variant |
|
|||||
Mucous membrane pemphigoid-like variant |
|
|||||
Linear IgA disease-like variant |
|
|||||
Brunsting-Perry pemphigoid-like variant |
|
|||||
Complications of cutaneous lesions:
Complications of mucosal lesions:
Diagnosis of epidermolysis bullosa acquisita is based on a combination of clinical and immunohistological findings:
An initial evaluation should include:
Further investigations to confirm the diagnosis include:
The goal of treatment for epidermolysis bullosa acquisita is to prevent disease progression and complications and to promote healing. Treatment should be tailored to the individual.
Patients should be advised to:
Multidisciplinary management (eg, gastroenterology, dentistry, and ophthalmology) is usually required where there is mucosal involvement.
Several treatment options are available, though there is no consensus on the most effective treatment due to the rarity of EBA.
Interventions reported in literature include:
Epidermolysis bullosa acquisita is a chronic, relapsing-remitting disease and those affected have a normal life expectancy. Prognostic factors are not fully understood, but prognosis is considered more favourable in children compared to adults.
Treatment aims to control the disease and often requires long-term maintenance therapy, which also poses health risks.

