Introduction
Demographics
Causes
Clinical features
Complications
Differential diagnoses
Diagnosis
Treatment
Outlook
Epidermolysis bullosa pruriginosa (OMIM 604129) is a rare and clinically heterogeneous form of dystrophic epidermolysis bullosa (dystrophic EB), resulting from a mutation in the type VII collagen gene.
First described in 1994, there have been fewer than 100 cases documented in the literature, and as such, little is known about the condition or its treatment.
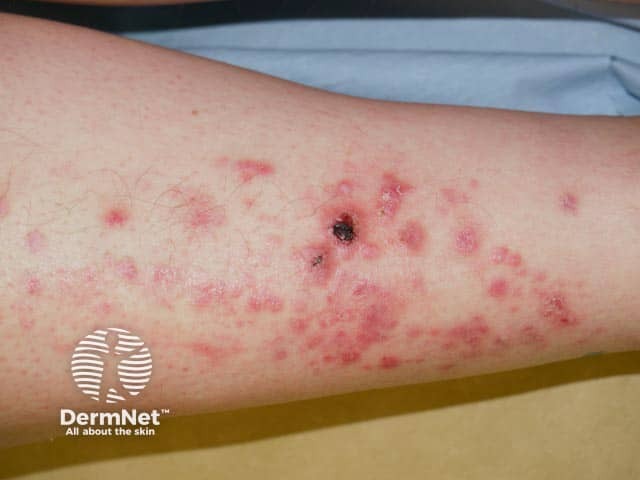
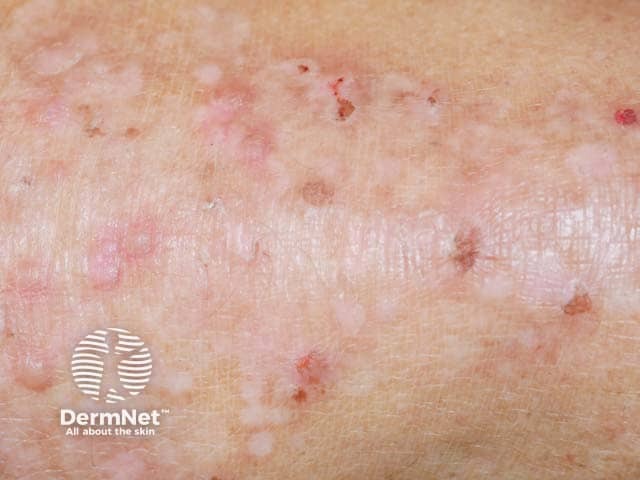
Epidermolysis bullosa (EB) pruriginosa can either present shortly after birth, or in adulthood.
As with other forms of dystrophic EB, the clinical findings of EB pruriginosa are attributed to mutations in the COL7A1 gene on chromosome 3p21.3, which encodes type VII collagen. Case reports of epidermolysis bullosa pruriginosa reveal many different alterations to this gene, including missense, nonsense, frame shift, and splice-site mutations.
Type VII collagen is a major skin structural component of the anchoring fibrils at the dermo-epidermal junction (DEJ). Mutations in the coding gene hamper the function of the anchoring fibrils and lead to a split in the DEJ below the level of the lamina densa. Clinically, this manifests as trauma-induced blisters, which heal leaving a scar.
Inheritance is variable; autosomal dominant and autosomal recessive patterns of inheritance have been reported. Most cases however have been found to be sporadic.
The characteristic pruritic component of the condition is poorly understood. A raised serum IgE level has been found in 7 of 9 examined patients with epidermolysis bullosa pruriginosa, and in some cases was found to be over 3 times the upper limit of normal. However, a personal or family history of atopy was a confounding factor in three of these patients, and so the results are far from conclusive.
EB pruriginosa is characterised by intensely itchy (pruritic) hypertrophic papules, nodules, and plaques usually on the lower extremities.
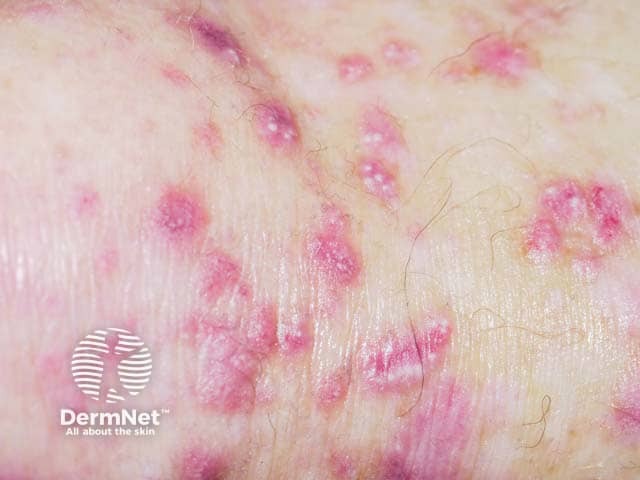
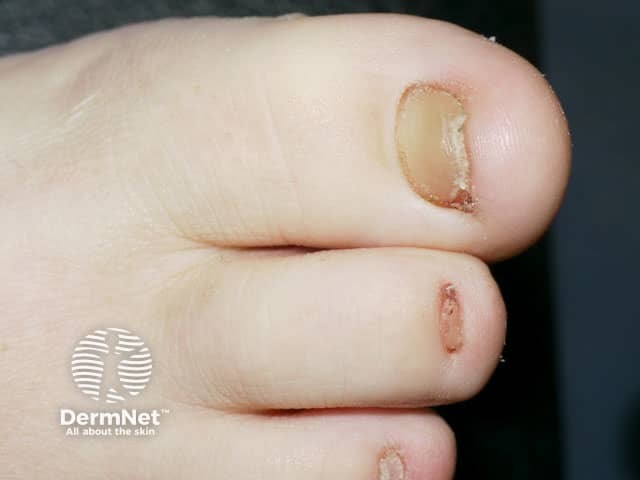
Epidermolysis bullosa pruriginosa should be considered clinically based on family history, clinical features, and a positive mechanobullous history, and confirmed with investigations.
Light microscopy of skin biopsy
Direct immunofluorescence on skin biopsy - negative
Electron microscopy
Genetic testing
Success has been varied and many treatments have been reported for a single case. Sustained responses have been recently reported with dupilumab.
Little is known about the long-term outcome for patients with EB pruriginosa. It is lifelong and often refractory to treatment.
No long term data exists with regards to the sustainability of medication-induced remission.

