

Amyloidosis cutis dyschromica — extra information
Introduction
Demographics
Causes
Clinical features
Complications
Diagnosis
Differential diagnoses
Treatment
Outcome
What is amyloidosis cutis dyschromica?
Amyloidosis cutis dyschromica is a form of primary cutaneous amyloidosis that causes localised hyperpigmentation and hypopigmentation.
Amyloidosis cutis dyschromica was first described by Japanese dermatologist Morishima in 1970 [1].
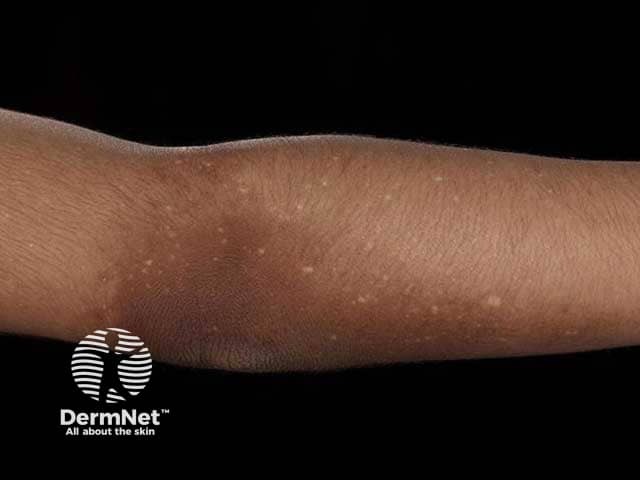
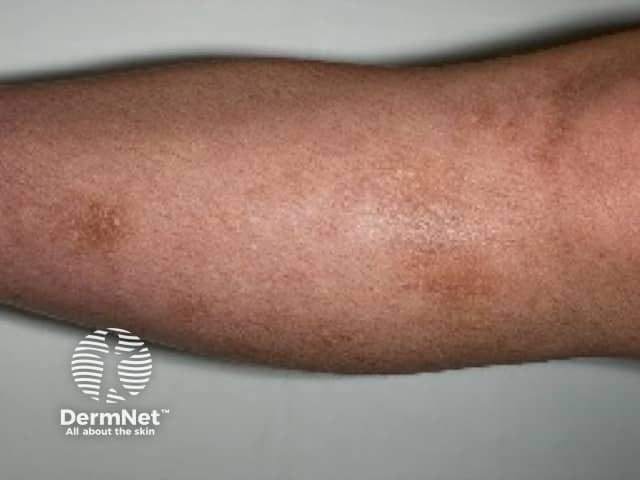
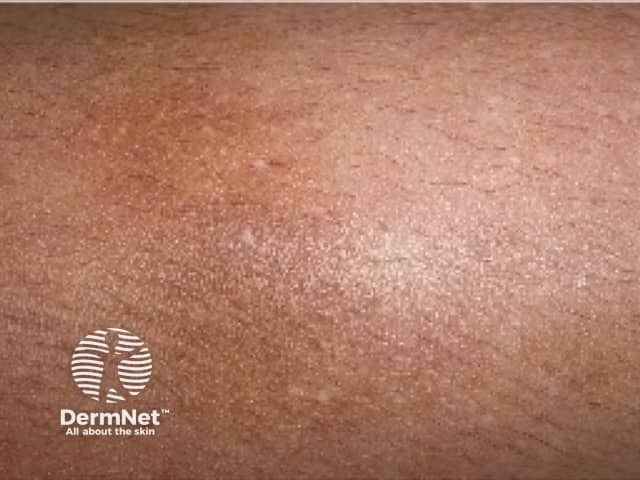
Who gets amyloidosis cutis dyschromica?
Amyloidosis cutis dyschromica is rare. At the time of writing, about 50 cases have been reported in the medical literature [2].
- Men and women are equally affected.
- Most patients first develop skin pigment changes in early childhood.
- The majority of patients (77%) have a family history of similar pigment alteration.
- Most cases (63%) have occurred in people of East Asian (Chinese, Hong Kong Chinese and Taiwanese) or South-East Asian (Thai, Filipino and Indonesian) ethnicity.
- Amyloidosis cutis dyschromica is also commonly seen in people of Indian and Pakistani heritage.
- Only three cases of amyloidosis cutis dyschromica have been reported in Caucasian patients.
What causes amyloidosis cutis dyschromica?
Amyloidosis cutis dyschromica is caused by the deposition of amyloid derived from degenerate or damaged keratinocytes. The reason why the keratinocytes degenerate and how this relates to the loss and gain of pigment in the affected areas of skin is not clear. A genetic cause is likely.
What are the clinical features of amyloidosis cutis dyschromica?
Amyloidosis cutis dyschromica causes slowly progressive localised hyperpigmentation and hypopigmentation (dyschromica or dyschromatosis).
- In most cases, the onset of the colour change is in childhood, although in some individuals the changes are not noted until adulthood.
- The dyschromica does not affect palms, soles or mucosal surfaces.
- Facial involvement is uncommon (approximately 10%) [2].
The specific findings that distinguish amyloidosis cutis dyschromica from the more common macular and lichenoid variants of primary cutaneous amyloidosis are:
- Dotted, reticular or diffuse hyperpigmentation mixed with ‘lentil-sized’ hypopigmented macules.
- Mild or no associated pruritus.
What are the complications of amyloidosis cutis dyschromica?
There are no known complications from amyloidosis cutis dyschromica.
How is amyloidosis cutis dyschromica diagnosed?
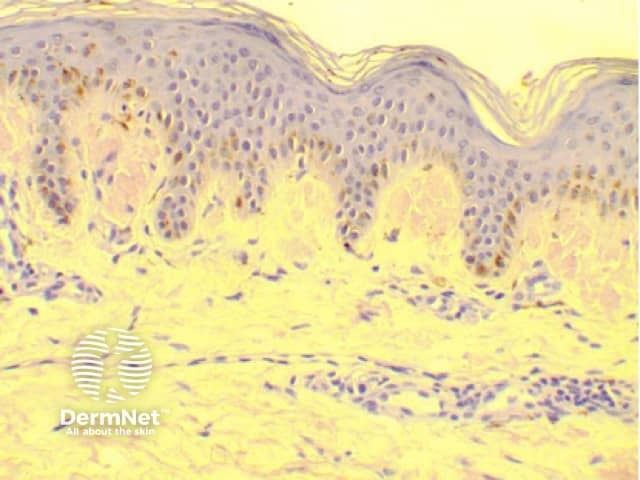
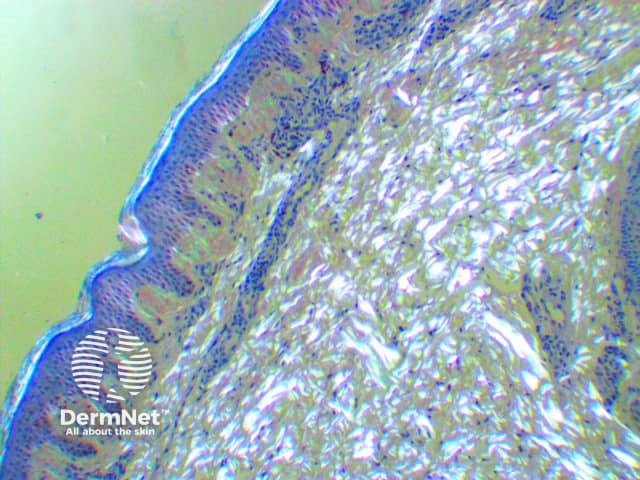
Amyloidosis cutis dyschromica is diagnosed by recognising the typical clinical features and confirmed by the biopsy of a hyperpigmented or hypopigmented macule.
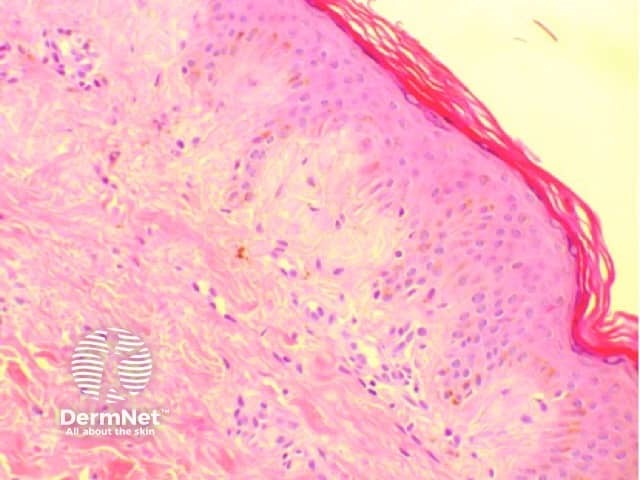
- Amyloid deposits are present within the papillary dermis just below the epidermis.
- They may be very subtle and difficult to see with haematoxylin and eosin (H&E) staining so Congo red staining is required.
- Using polarised light, apple-green-coloured birefringence of the amyloid material may be seen on microscopic examination.
Histology of amyloidosis cutis dyschromica
What is the differential diagnosis for amyloidosis cutis dyschromica?
Amyloidosis cutis dyschromica may closely resemble a number of other rare dyschromatoses; these include:
- Dyschromatosis universalis hereditaria
- Dyschromatosis symmetrica hereditaria
- Cutis tricolor
- Epidermolysis bullosa simplex with mottled pigmentation
- Tertiary pinta.
Other conditions that should be excluded include:
- Dyskeratosis congenita, which presents with nail dystrophy, oral leukoplakia, and bone marrow failure in childhood
- Xeroderma pigmentosum, which causes marked cutaneous and ocular photosensitivity and childhood-onset aggressive skin cancers
- Degos disease, a potentially life-threatening vasculopathy.
What is the treatment for amyloidosis cutis dyschromica?
No therapeutic intervention has been demonstrated to be of benefit in amyloidosis cutis dyschromica. Multiple topical treatments, including 10% urea cream and tazarotene [1], have been prescribed to patients without significant improvement. Oral vitamin C and vitamin E supplements have had minimal benefit [3]. Acitretin has been prescribed in a small number of cases, with a modest improvement reported in approximately 85% of cases [2].
What is the outcome for amyloidosis cutis dyschromica?
Amyloidosis cutis dyschromica is typically gradually progressive. The dyschromia may eventually involve almost all of the skin except the palms, soles of the feet, and oral and genital mucosa. Alternatively, amyloidosis cutis dyschromica can progress gradually in localised areas, and it is often more prominent in areas of skin overlying bony prominences and joint surfaces.
References
- Morishima T. A clinical variety of localized cutaneous amyloidosis characterized by dyschromia (amyloidosis cutis dyschromica). Jpn J Dermatol Series B 1970; 80: 43–52.
- Mahon C, Oliver F, Purvis D, Agnew K. Amyloidosis cutis dyschromica in two siblings and review of the epidemiology, clinical features and management in 48 cases. Australas J Dermatol 2016; 57: 307–11. DOI: 10.1111/ajd.12342. PubMed
- Qiao J, Fang H, Yao H. Amyloidosis cutis dyschromica. Orphanet J Rare Dis 2012; 7: 95. DOI: 10.1186/1750-1172-7-95. PubMed Central