

Epidermolysis bullosa — extra information
Introduction
Demographics
Clinical features
Diagnosis
Assessment of severity
Treatment
What is epidermolysis bullosa?
Epidermolysis bullosa (EB) is a group of inherited diseases that are characterised by blistering lesions on the skin and mucous membranes. These may occur anywhere on the body but most commonly appear at sites of friction and minor trauma such as the feet and hands. In some subtypes, blisters may also occur on internal organs, such as the oesophagus, stomach and respiratory tract, without any apparent friction.
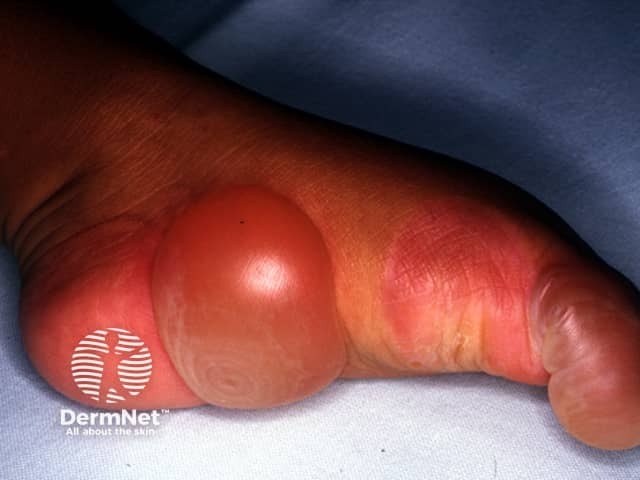
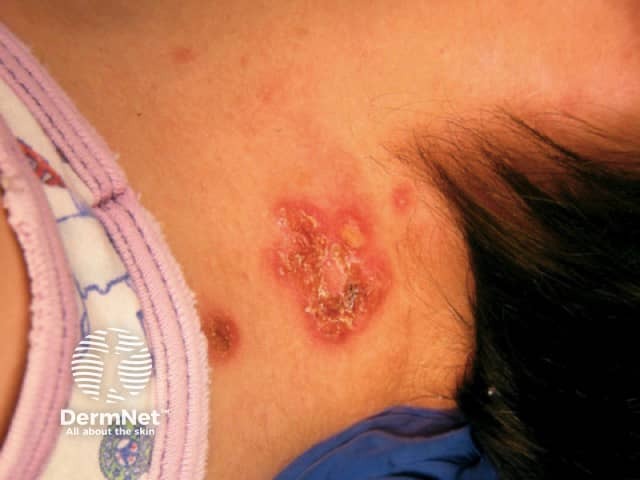
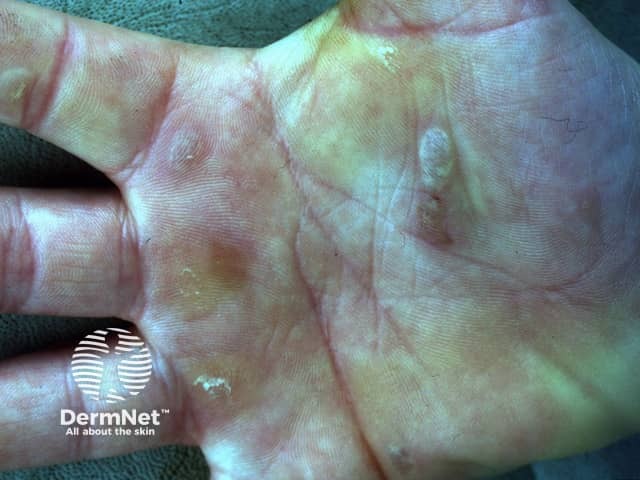
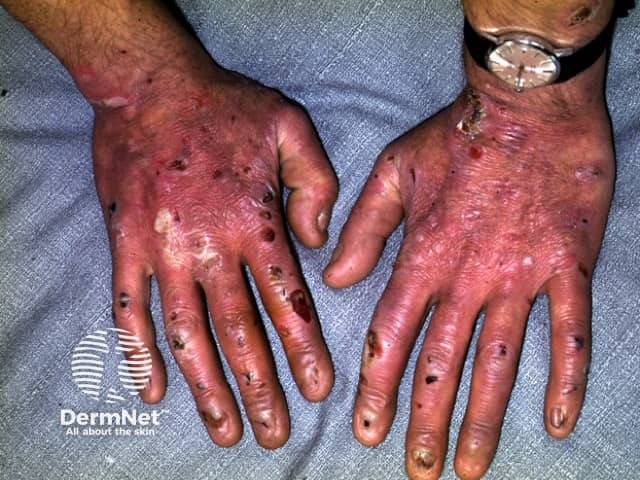
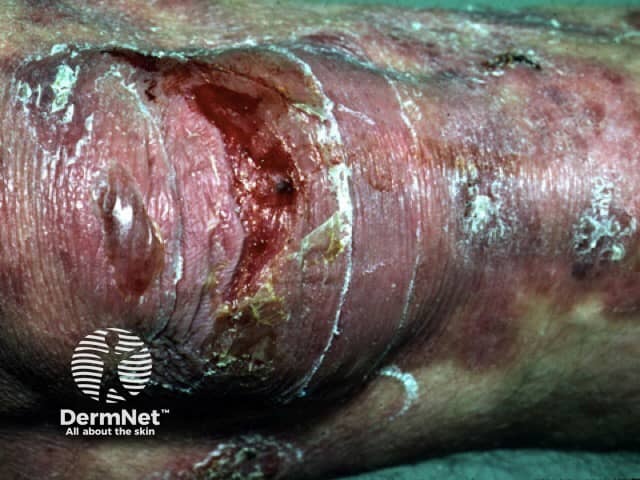
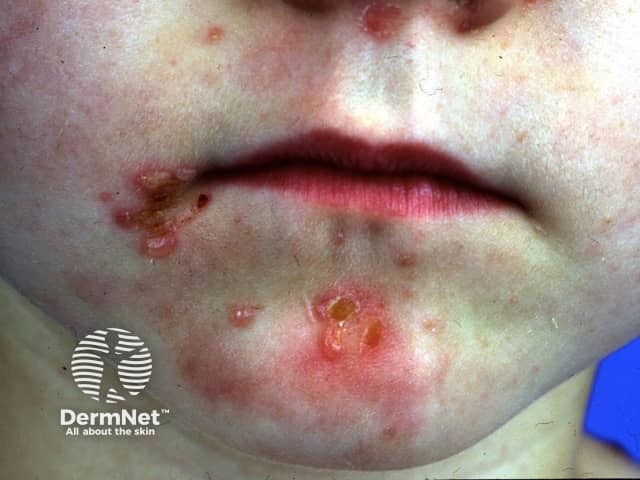
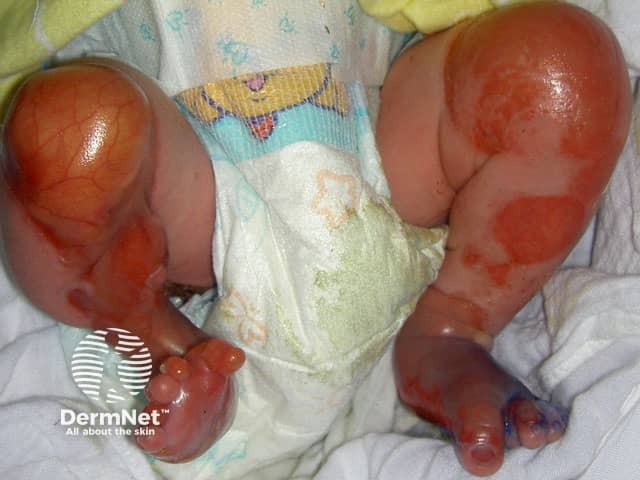
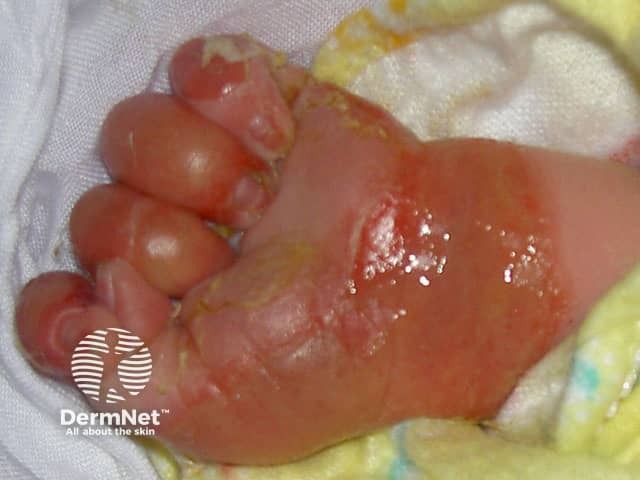
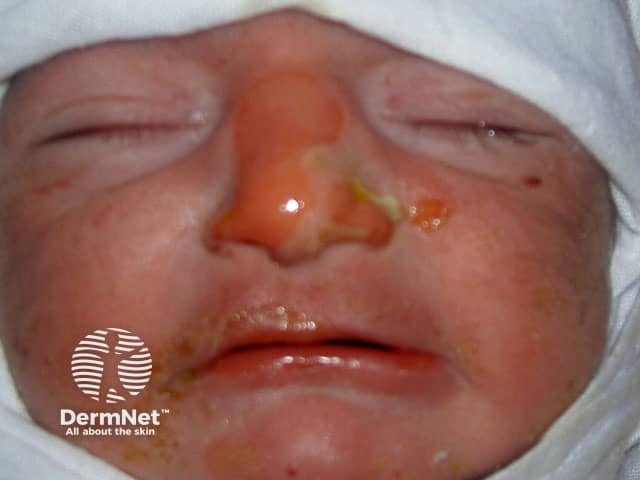
EB should be distinguished from common friction blisters, and from epidermolysis bullosa acquisita (EBA), which is a blistering autoimmune disease that is not inherited and often doesn't develop until adult life.
The EB conditions result from genetic defects of molecules in the skin concerned with adhesion. Loss of adhesion results in blister formation. There are four major types of EB based on different sites of blister formation within the skin structure:
EB type |
The site of blister formation within skin |
|---|---|
Epidermis or uppermost layer of skin cells (keratinocytes) |
|
Lamina lucida within the basement membrane zone |
|
Lamina densa and upper dermis |
|
Mixed pattern or multiple levels within and beneath the basement membrane zone |
Within each of these types of EB, there are various subtypes. Varying degrees of severity that range from mild to severe are found with each EB type. International consensus on the diagnosis and classification of EB has resulted in updated recommendations (2014), based on newer clinical and molecular data.
Who gets epidermolysis bullosa?
EB is an inherited disease, which means that you have inherited one or two EB genes. In autosomal dominant EB, only one abnormal gene is needed to express the disease. This means only one parent needs to carry the EB gene. On the other hand, autosomal recessive inherited EB requires you to have two EB genes (one from each parent) to have the disease. If a person has one recessive EB gene paired with a normal gene they are called a carrier and do not have the disease.
EB usually occurs at birth or shortly after. Males and females are equally affected. Occasionally EB may be mild enough at birth not to be apparent, and it is not until the child is older or reaches adulthood before it is detected.
What are the clinical features of epidermolysis bullosa?
Epidermolysis bullosa simplex (EBS)
For more details, see Epidermolysis bullosa simplex.
EBS Subtypes |
Features |
|---|---|
Localised EBS Previously known as Weber-Cockayne |
|
Generalised EBS Previously known as Koebner |
|
Generalised severe EBS Previously known as Dowling Meara |
|
Junctional epidermolysis bullosa (JEB)
For more details, see Junctional epidermolysis bullosa.
JEB Subtypes |
Features |
|
|---|---|---|
Generalised severe JEB Previously known as Herlitz |
|
|
Generalised intermediate JEB Previously known as Non-Herlitz |
|
|
Dystrophic epidermolysis bullosa (DEB)
For more details, see Dystrophic epidermolysis bullosa.
DEB Subtypes |
Features |
|
|---|---|---|
Dominant generalised DEB |
|
|
Generalised severe recessive (R) DEB Previously known as Hallopeau-Siemens; and; Generalised intermediate RDEB (previously Non-Hallopeau-Siemens) |
|
|
Kindler syndrome
Kindler syndrome |
Features |
|
|---|---|---|
Kindler syndrome |
|
|
There are many other subtypes of EB. The presentation and severity of EB are affected by the specific genetic changes and can at times be difficult to classify.
Genetic counselling and prenatal testing is available for EB in some centres and should be considered.
Referral for psychological support should also be considered for children with EB and their family, especially for the more severe subtypes.
How is epidermolysis bullosa diagnosed?
In the dominant subtypes of EB, where an informative family tree is known, it is often acceptable for a clinical diagnosis (based on the presenting signs) to be made by a specialist dermatologist.
Diagnostic tests are also available in some countries and include skin biopsy of a newly induced blister which undergoes immunofluorescence antigen mapping (IFM) and transmission electron microscopy (EM). Mutational analysis (blood testing of genes), although not currently considered the first-line diagnostic test, is also available in some countries.
A referral to a dermatologist will be required.
How is the severity of epidermolysis bullosa assessed?
The severity of EB can be assessed using the following scoring systems:
- The Birmingham EB score
- The EBDASI: EB Activity and Scarring Index
- The iSCOREB: an instrument for scoring clinical outcomes for research of EB
- The QOLEB: Quality of life evaluation in EB
Treatment of epidermolysis bullosa
General
There is no cure for EB. However, significant research, including gene therapy and cell-based therapy, continue in the aim to improve quality of life.
Most current treatment is symptomatic. The primary aim is to protect the skin and stop blister formation, promote healing, and prevent complications.
Because EB can affect so many different parts of the body, a team of medical specialists is usually required for overall care. When necessary, treatment with oral and topical medications may be prescribed by your doctor to assist healing or prevent complications.
The following are some general measures used in caring for a patient with EB.
- Avoid activities that induce friction on the skin. This includes the handling of infants and children – alternative handling techniques are easily learnt from a trained health care professional.
- Maintain a cool environment and avoid overheating
- Use foam padding or sheepskins to help reduce friction on pieces of furniture such as beds, chairs and infant car seats
- Choose clothing (including nappies) and footwear that is light, has no irritating seams or detail, (eg, zips and tight elastic).
- Pierce, drain and dress blisters to promote healing (this should be done only by people who have received training on wound care)
- Many traditional adhesive tapes and dressings may be unsuitable for people with EB — especially those with the more severe forms (eg, RDEB) — as their removal can cause additional trauma to the skin. Use of advanced wound care products such as low-adherent silicone tapes and dressings is recommended. However, resourcefulness by using items readily available, such as applying additional lubrication (eg, Vaseline/paraffin oil) to some traditional wound dressings, is helpful.
- Oleogel-S10, containing birch triterpenes or birch bark extract, has been shown to accelerate the healing of chronic wounds in junctional and dystrophic epidermolysis bullosa.
- When EB affects other parts of the body, various cares and treatments are adopted. For example, a soft diet when the oesophagus is involved or using stool softeners for constipation, or if the patient has anal blisters.
- Clinical practice guidelines are available on the DEBRA* International website including those on Oral Health care, Wound Care, Pain Management, Cancer Management.
- Further support and information from both health care professionals and those affected by EB:
- DEBRA (Dystrophic Epidermolysis Bullosa Research Association) International has over 50 member groups worldwide.
- Eb-Clinet is a clinical network or EB centres and experts across the world.
References
- OMIM — Online Mendelian Inheritance in Man (search term: Epidermolysis bullosa)
- Dermatologic Clinics Volume 28, Issue 1, Pages 1–196 (January 2010) Epidermolysis Bullosa: Part I — Pathogenesis and Clinical Features. Journal
- Dermatologic Clinics Volume 28, Issue 2, Pages 197–452 (April 2010) Epidermolysis Bullosa: Part II — Diagnosis and Management. Journal
- El Hachem M, Zambruno G, Bourdon-Lanoy E, et al. Multicentre consensus recommendations for skin care in inherited epidermolysis bullosa. Orphanet J Rare Dis 2014; 9: 76. doi: 10.1186/1750-1172-9-76. PubMed PMID: 24884811; PubMed Central
- Fine JD, Bruckner-Tuderman L, Eady RA, et al. Inherited epidermolysis bullosa: updated recommendations on diagnosis and classification. J Am Acad Dermatol 2014; 70: 1103–26. doi: 10.1016/j.jaad.2014.01.903. PubMed
- Kern JS, Sprecher E, Fernandez MF, et al. Efficacy and safety of Oleogel-S10 (birch triterpenes) for epidermolysis bullosa: results from the phase III randomized double-blind phase of the EASE study. Br J Dermatol. 2023;188(1):12-21. doi:10.1093/bjd/ljac001 Journal
On DermNet
- Epidermolysis bullosa simplex
- Junctional epidermolysis bullosa
- Dystrophic epidermolysis bullosa
- Kindler syndrome
- Epidermolysis bullosa pruriginosa
- Epidermolysis bullosa acquisita
- Epidermolysis bullosa with congenital absence of skin
- Friction blisters
- Blistering diseases
- Prenatal testing of inherited skin disorders
- Blistering skin conditions
- Blisters and pustules in neonates
- Cockayne syndrome
Other websites
- Blistering disorders — DermNet e-lectures [Youtube]
- EB-CLINET — a clinical network of EB centres and experts
- DEBRA International
- DebRA Dystrophic EB Research Association of New Zealand
- DebRA of America Inc.
- DEBRA UK
- Australasian Blistering Diseases Foundation (includes guidelines for referring a biopsy for diagnosis at the National EB Diagnostic Lab in Australia)
- Asociacion de Epidermiolisis Bulosa de Chile
- Epidermolysis bullosa — Medscape
- Epidermolysis bullosa with pyloric atresia — US National Library of Medicine Genetics Home Reference
- Dystrophic epidermolysis bullosa — US National Library of Medicine Genetics Home Reference
- Junctional epidermolysis bullosa — US National Library of Medicine Genetics Home Reference
- Epidermolysis bullosa simplex — US National Library of Medicine Genetics Home Reference
- Dystrophic Epidermolysis Bullosa — British Association of Dermatologists
- Epidermolysis Bullosa Simplex — British Association of Dermatologists
- Epithelial Adhesion — geneSkin
- GeneTests — a medical genetics information resource