Introduction Demographics Causes Clinical features Complications Diagnosis Differential diagnoses Treatment Outcome
Fabry disease is a rare inherited lysosomal storage disorder [1]. It is also known as Anderson–Fabry disease and angiokeratoma corporis diffusum.
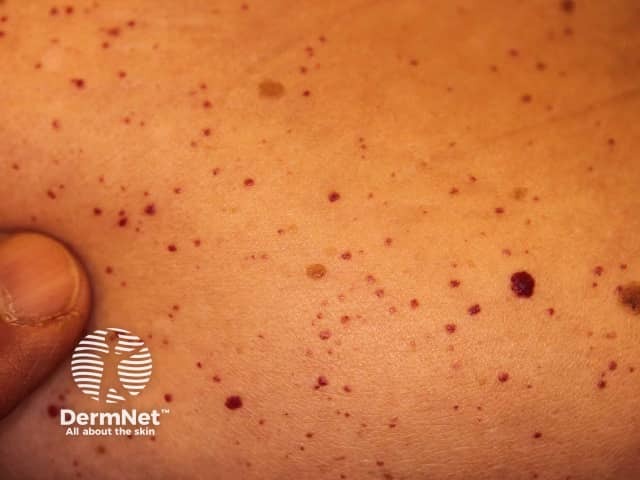
Fabry disease causes clusters of angiokeratomas (small, dark red spots on the skin) and many systemic symptoms due to the deposition of globotriaosylceramide (Gb3) in multiple organs.
The estimated prevalence in males is about one in 40,000–60,000 of the population [2]. Fabry disease generally affects men and boys more severely and at an earlier age than women and girls because its inheritance is X-linked (the male sex only carries one X chromosome whereas females have two). Fabry disease can occur in all ethnic groups [3].
The age of onset of Fabry disease can range from the second to the fifth decade of life or later.
Fabry disease often presents with non-specific symptoms that can be mild and subtle, and it is commonly missed or misdiagnosed, leading to an underestimation of its prevalence [4].
Fabry disease is caused by a mutation of the alpha-galactosidase A gene (GLA) mapped to the long arm of the X chromosome [5]. There are hundreds of different pathogenic variants of the mutation, resulting in differing symptoms and variable severity [6]. Men with Fabry disease pass the gene on to all daughters but to none of their sons. Women with Fabry disease have a 50% chance of passing the gene onto their daughters or to their sons. Some women and girls are affected as severely as men and boys due to a random X chromosome inactivation [7].
Gb3 is an intermediate in the degradative pathway of globoside (a glycosphingolipid), a major component of some cell membranes. The enzyme alpha-galactosidase A catalyses the cleavage (separation) of the terminal galactose from Gb3. Lack of the alpha-galactosidase A enzyme leads to accumulation of Gb3 in various tissues, leading to cell death. The most clinically significant sites of accumulation of Gb3 are blood vessels of the skin, heart, nerves, and kidneys [8].
The characteristic features of Fabry disease are:
Dermatological manifestations occur in more than 70% of patients with Fabry disease, with a mean age of onset at 17 years [3].
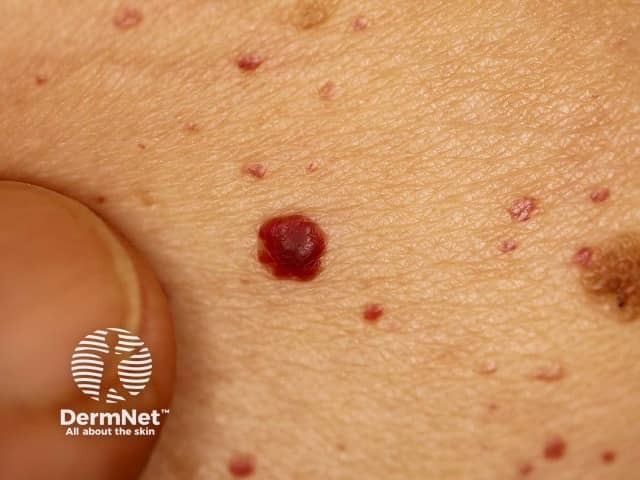
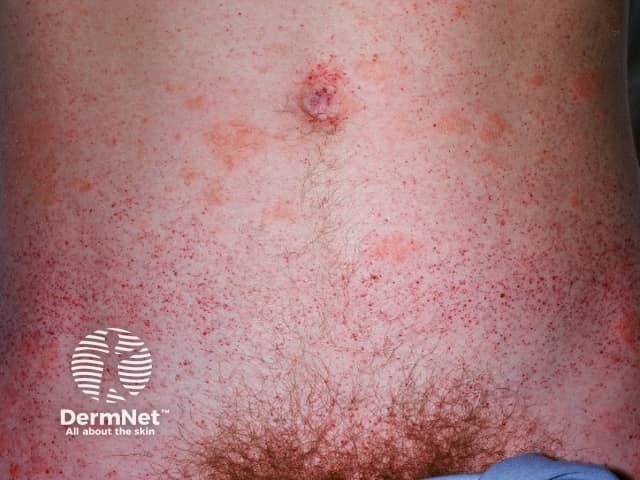
Angiokeratomas are dilated blood vessels in the upper dermis, presenting as red or black papules. The papules do not blanch with pressure, and older lesions can become warty. Clusters or diffuse angiokeratomas first appearing in young adults must alert physicians to a possible diagnosis of Fabry disease.
In Fabry disease, angiokeratomas are caused by the accumulation of Gb3 in the dermal endothelial cells, which leads to bulge and incompetence of the vessel walls [12]. The angiokeratomas usually localise in the bathing suit area (from the umbilicus to the upper thighs, including the genitals). More than one-third of patients also develop angiokeratomas and telangiectasia on the lips and inside the mouth [13].
There is no correlation between the severity of the disease and the extent of angiokeratomas [14].
Hypohidrosis is a common feature of Fabry disease that leads to dry skin and heat intolerance [15]. It is likely the result of an abnormal autonomic nerve response due to Gb3 deposition.
Fabry disease is identifiable by other skin signs, such as:
The three most serious complications of Fabry disease are kidney disease, cardiac disease, and cerebrovascular disease [3].
Male and female patients are diagnosed differently.
Family members of the diagnosed patient should also be tested for Fabry disease.
Urinalysis and ECG should be undertaken to detect renal and cardiac abnormalities.
Due to its rarity and the wide range of non-specific clinical features, Fabry disease is often misdiagnosed, and is colloquially considered a “great imposter” condition [16].
Angiokeratomas are not characteristic of Fabry disease as solitary lesions can occur in healthy individuals. They are also found in patients with hereditary haemorrhagic telangiectasia and in the other lysosomal disorders, including:
The most commonly mistaken diagnoses are the following:
Once diagnosed, male patients with Fabry disease are often prescribed enzyme replacement therapy, regardless of the clinical features [9,14,18]. Female patients should only receive treatment if they have significant clinical manifestations.
Enzyme replacement therapy provides patients with the deficient enzyme alpha-galactosidase A. It reduces the severity and progression of disease manifestations, especially neuropathic pain and kidney disease, by reducing the tissue deposition of Gb3. The effect of enzyme replacement therapy on cardiovascular manifestations is less clear [18].
Patients with Fabry disease may require input from different specialities input including from GPs, cardiologists, nephrologists, neurologists, and dermatologists to manage their individual organ manifestations. Neuropathic pain of the hand and feet may respond to anticonvulsants, such as carbamazepine [18,19].
Treatment for angiokeratomas includes cryotherapy, electrocoagulation, excisional surgery, and laser therapy [12,14,20].
Fabry disease is progressive and usually results in a shortened life expectancy. The prognosis varies from patient to patient [21]. Survival is significantly reduced in male patients with severe forms of Fabry disease to below 60 years, often to 40 or 50 years. Most deaths are due to cardiovascular disease including stroke, myocardial infarction, and heart failure. Death from renal failure, sepsis, and suicide have also been reported [18]. The benefit of enzyme replacement therapy on overall prognosis is unclear due to a lack of current evidence [18,22,23].

