Introduction Demographics Causes Classification Clinical features Complications Diagnosis Differential diagnoses Treatment Outcome
Hereditary angioedema is a familial disease characterised by recurrent attacks of self-limiting oedema. It can affect the skin, gastrointestinal tract, and airways.
Hereditary angioedema is an autosomal-dominant condition, meaning if one parent has the abnormal gene that codes for angioedema, half of their children will inherit the condition. Around 25% of cases are due to spontaneous mutations.
The prevalence of hereditary angioedema is estimated at 1 in 50,000 persons. There is no particular prevalence amongst ethnic groups or between sexes.
The gene coding for C1-INH is located on chromosome 11 and nearly 200 mutations of this gene have been described.
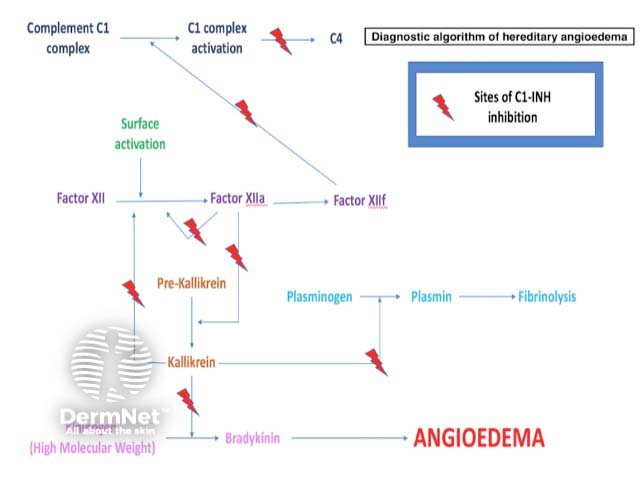
The most common type of hereditary angioedema is associated with a deficiency of functional C1-esterase inhibitor (C1-INH), a serine protease inhibitor that regulates the activation of the classic complement pathway and suppresses spontaneous activation of complement component C1 (figure 1).
C1-INH also regulates the contact (kallikrein-kinin) system, which is responsible for inflammation and blood coagulation. It involves factor XII, factor XIIa, kallikrein, and high-molecular-weight kininogen. The activation of these molecules generates bradykinin, a peptide which promotes inflammation. A deficiency of functional C1-INH results in excessive bradykinin.
Bradykinin can increase vascular permeability by binding to its receptor on vascular endothelial cells, causing angioedema.
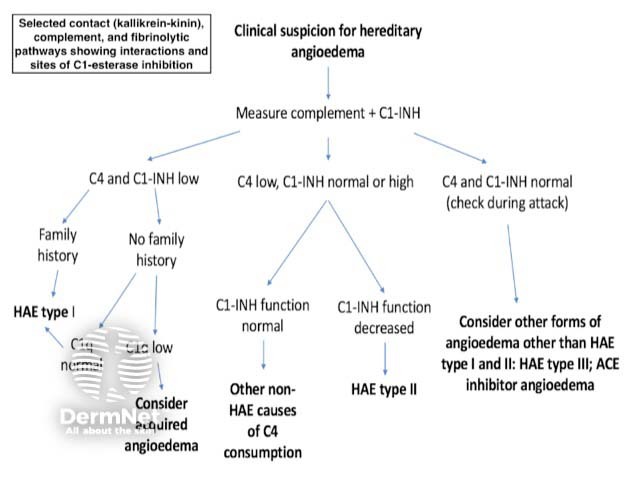
Hereditary angioedema is classified into three types based on pathogenesis (figure 2).

Symptoms of hereditary angioedema typically begin in childhood, worsen in puberty, and persist throughout life.
Untreated patients have attacks every 7–14 days on average, with a range from every 3 days to almost never.
One-third of patients with hereditary angioedema experience early symptoms of erythema marginatum that may be confused for urticaria. Erythema marginatum is a non-pruritic form of annular erythema more commonly associated with rheumatic fever. The lesions can be static or extend peripherally in a centrifugal way.
The angioedema generally increases over the first 24 hours and then subsides over the next 2–5 days. Oedema may affect an arm, leg, hand, foot, or the abdomen. Oropharyngeal swelling is less common. One or all of these areas can be affected in an attack and it may be asymmetrical.
Stress and trauma, including surgery, can be triggers for episodes, but most episodes of hereditary angioedema are spontaneous.
An abdominal attack of hereditary angioedema can cause severe abdominal pain, nausea, and vomiting; it is often mistaken for an 'acute abdomen'. Bowel sounds may be absent, and guarding (muscle spasms) may be present on examination. Therefore, many patients with hereditary angioedema undergo unnecessary abdominal surgery, especially prior to the diagnosis of hereditary angioedema.
Laryngeal oedema can be fatal. Historical data reports asphyxiation and death occurring in over 30% of patients with hereditary angioedema without treatment.
Angioedema symptoms are often disabling and disfiguring. Patients may suffer from depression, anxiety, impaired function, and reduced quality of life.
Hereditary angioedema is a rare disease and it is therefore encountered infrequently by physicians. This may lead to a delay in diagnosis. The clinician should consider a diagnosis of hereditary angioedema in patients with recurrent attacks of angioedema in the absence of urticaria.
Investigations should include complement levels (figure 3).
The main differential diagnoses for hereditary angioedema are:
Acquired angioedema results from a lymphoproliferative disorder such as a B-cell lymphoma (type I) or from the development of mainly immunoglobulin G antibodies against C1-INH (type II). C1q measurement can help to distinguish acquired angioedema from hereditary angioedema. C1q levels are normal in hereditary angioedema but low in acquired angioedema, which otherwise has a similar C1-INH/C4 profile.
Acute allergic angioedema occurs in response to a known trigger that provokes an associated histamine response with urticaria and pruritus. Antihistamines are helpful in this condition where they afford no benefit in hereditary angioedema. On investigation, serum complement levels are normal as opposed to low in hereditary angioedema.
ACE inhibitor-induced angioedema occurs because the inhibition of ACE allows bradykinin to accumulate. Serum complement levels are normal.
Chronic spontaneous urticaria with angioedema is classified as an autoimmune disorder. Serum complement levels are normal.
Patients with hereditary angioedema should be counselled to avoid agents that may precipitate attacks such as an ACE inhibitor or oestrogen. Stress is thought to be a trigger for attacks, so stress reduction or management may also be advised.
Treatment is divided into three areas:
Long-term prophylaxis is indicated for patients with frequent or severe attacks. The options include:
Androgens are contraindicated in pregnancy, lactation, cancer, hepatitis, and childhood. Side effects of androgens include virilisation, weight gain, acne, hypertension, and atherosclerosis. The lowest possible dose should be used to control hereditary angioedema to minimise side effects.
The mechanism of action of tranexamic acid in hereditary angioedema is unknown. Side effects included nausea, diarrhoea, myalgia, and thrombosis.
Short-term prophylaxis is indicated for patients who are undergoing planned surgical or dental procedures.
Novel targeted therapies such as a plasma-derived C1-esterase inhibitor, a kallikrein inhibitor (ecallantide), or a BDKRB2 antagonist (icatibant) should be given prior to the procedure if available.
Alternatives are a 5-day course of danazol or tranexamic acid before the procedure.
A plasma-derived C1-esterase inhibitor, a kallikrein inhibitor, or a BDKRB2 antagonist should be administered as soon as possible after an acute attack if available.
Supportive care, including fluid replacement and pain management, should also be implemented.
The recent development of novel targeted therapies has led to improved morbidity and mortality for patients with hereditary angioedema.

