Introduction
Demographics
Causes
Clinical features
Complications
Diagnosis
Differential diagnoses
Treatment
Outcome
Behçet disease (BD) is a rare, multi-system condition characterised by recurrent painful oral and genital ulcers, a variety of skin lesions, and eye problems.
Many other organs such as the gut, nervous system, lungs, and arteries may also be affected. It is a form of vasculitis which primarily targets small arteries, but can affect both arteries and veins of all sizes.
It is also known as Adamantiades-Behçet disease, Behçet syndrome, and malignant aphthosis.
See more Behçet disease images
Behçet disease is most common in individuals originating from eastern and central Asia as well as the eastern Mediterranean. It is most prevalent in Turkey (80–370 cases per 100,000), followed by Iran, and Japan. It is much rarer in those of northern European, African, and Latin American descent.
Onset is usually between 30 and 40 years of age. It rarely occurs in children. It was previously thought that BD was more common in males compared with females, but more recent data suggests that incidence is similar, although this varies between different ethnic and geographic groups.
Behçet disease is of unknown cause, but it is thought to have an autoimmune basis. It is associated with several genetic factors, particularly HLA-B51. Several infections have also been implicated as potential triggers including Streptococcus sanguis, herpes simplex virus, hepatitis viruses, and parvovirus B19.
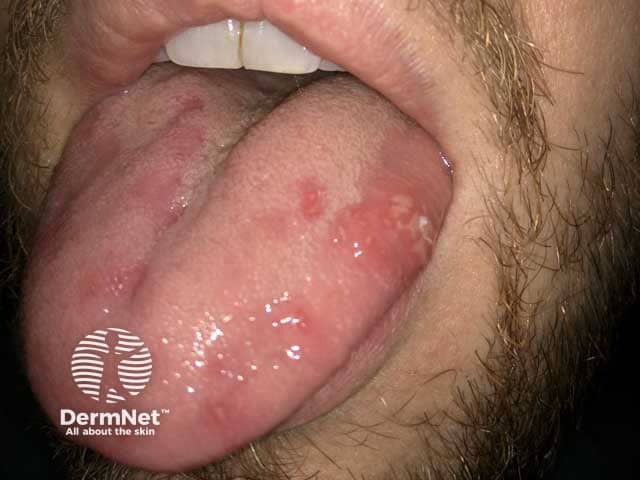
Oral ulcers — anywhere in the mouth, pharynx, and tonsils.
Genital ulcers — painful, recurrent ulcers on the vulva, vagina, perineum and inguinal areas, scrotum, and penis.
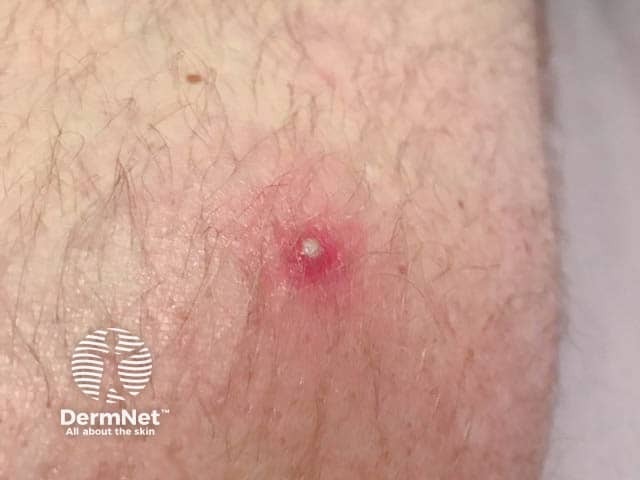
Pathergy is a commonly observed feature in Behçet disease, and refers to the eruption of papulopustules at the site of a needle injury within 24–48 hours. Pathergy is not specific to BD, and can also occur in pyoderma gangrenosum and Sweet syndrome.
Other skin lesions include:
For more images, see the Behçet disease image page.
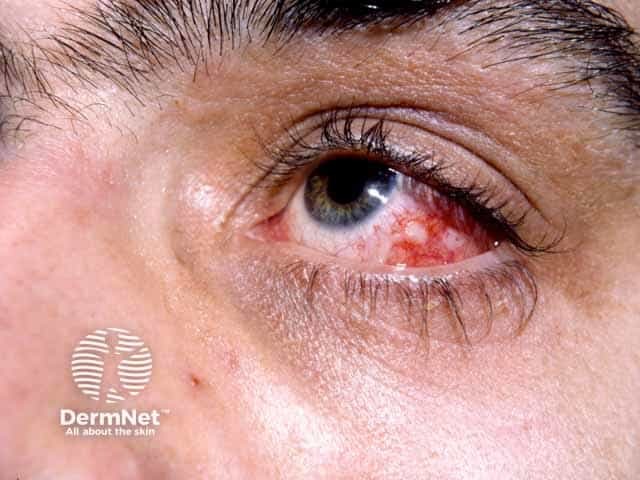
Ophthalmic lesions — seen in 70% of patients, and include:
Musculoskeletal symptoms include:
Vascular involvement is present in 25% of patients, resulting in:
Involvement of the aorta and pulmonary artery can cause life-threatening occlusion or haemorrhage from aneurysm formation.
Other organ systems which can be affected less commonly include neurological, pulmonary, renal, and cardiac.
The features are similar, although Behçet disease is less common in those with skin of colour.
Given the multi-system nature of Behçet disease, there are many potential complications.
The most serious complications include:
BD can be challenging to diagnose, and several years may elapse before a confident diagnosis can be made, and strict diagnostic criteria fulfilled. It is sometimes useful to classify sufferers as having possible, probable, or definite Behçet disease as new symptoms evolve. There are no definitive confirmatory diagnostic tests.
The International Study Group Criteria are widely used in diagnosis, and criteria include:
Common investigations include:
Other investigations may be required and depend on the organ system affected.
This depends on the clinical presentation but other common conditions which cause orogenital aphthous ulcers should be considered.
Differentials include:
Behçet disease treatment should be tailored to each individual patient based on clinical manifestations and extent of disease — an MDT approach is required. Behçet disease typically has a relapsing-remitting course, and treatments should aim to suppress any inflammatory exacerbations and prevent irreversible organ damage, particularly in the initial phase of disease.
Ocular, vascular, and neurological manifestations are often more serious and require more aggressive treatment.
Topical treatments include:
Systemic treatments for mucosal disease include:
With the exception of erythema nodosum and pyoderma gangrenosum, most other cutaneous lesions respond well to the above treatments. Recalcitrant acneiform lesions may require oral isotretinoin.
Erythema nodosum in Behçet disease:
Evidence of ulceration or detection of medium-vessel vasculitis on skin biopsy is an indication for systemic glucocorticoids with another immunosuppressive agent (typically azathioprine).
Pyoderma gangrenosum in BD should be managed as in other causes of this disease.
The key management points are:
Behçet disease typically runs a relapsing-remitting course and the prognosis varies depending on the organ systems affected.
Mortality is low but is increased in patients with neurological, vascular, and gastrointestinal involvement. Ophthalmic involvement is a major cause of morbidity.
The prognosis is worse in males.

