What is amyloidosis?
Amyloidosis is the term used for a group of diseases where various insoluble proteins (amyloid) accumulate in one or more body organs causing dysfunction of the organ system. Organs often affected include the heart, kidney, gastrointestinal tract, nervous system, and skin.
Amyloidosis of the skin is called cutaneous amyloidosis. In this condition, amyloid or amyloid-like proteins are deposited in the dermis.
Types of amyloidosis
There are three important types of amyloidosis.
- Localised amyloidosis — primary cutaneous amyloidosis is discussed below
- Systemic amyloidosis
- Familial (hereditary) amyloidosis:
- A rare inherited form, most commonly causing cardiomyopathy and neuropathy in middle age
- The most common form, known as ATTR amyloidosis, is due to a mutation in the transthyretin (TTR) gene on chromosome 5 and leads to faulty hepatic transthyretin protein.
What is primary cutaneous amyloidosis?
Primary cutaneous amyloidosis (PCA), also termed primary localized cutaneous amyloidosis, encompasses a group of skin conditions characterised by extracellular deposition of heterogenic amyloid protein in previously normal skin, without internal organ involvement. It is distinct and unrelated to systemic amyloidosis. Based on morphology and histology, PCA is subclassified into:
- Lichen amyloidosis (LA)
- Macular amyloidosis (MA)
- Biphasic amyloidosis (BA) which implies the coexistence of both lichen and macular amyloidosis
- Nodular amyloidosis (NA).
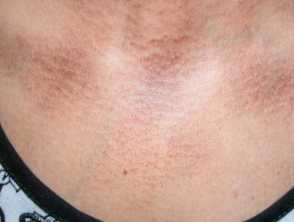
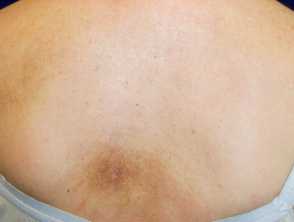
Rippled pigmentation on the upper chest due to macular amyloidosis
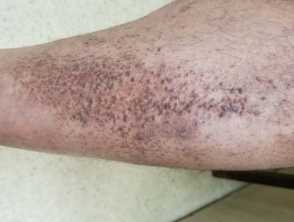
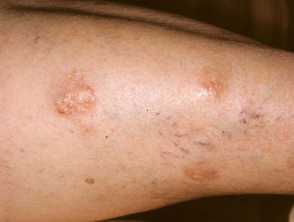
Scaly tan brown papules, some arranged lineraly in lichen amyloidosis on the leg
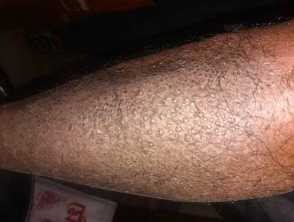
Extensive and confluent lesions of lichen amyloidosis on the shin
Macular amyloidosis showing rippled pigmetation over the upper back
Yellow waxy nodules of amyloidosis on the thigh
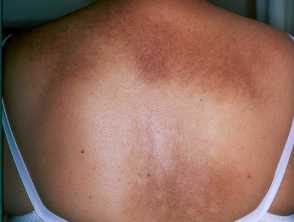
Repetitive scratching of an area of notalgia paraesthetica resulting in macular amyloidosis on the back
Primary cutaneous amyloidosis
There are also several rare atypical variants, such as:
- Amyloidosis cutis dyschromica
- Poikiloderma-like amyloidosis
- Bullous lichen amyloidosis
- Incontinentia pigmenti — like macular amyloid.
Who gets primary cutaneous amyloidosis?
- Primary cutaneous amyloidosis occurs more commonly in skin phototype III–IV people from Asia, South America, and the Middle East.
- It is rare in those with European origins (Caucasian). Lichen amyloidosis is highly prevalent in the South China region and Southeast Asia, and it is also more common than other subtypes worldwide. The biphasic form represents 15% of patients with PCA.
- Nodular amyloidosis is much rarer but attracts more investigation via biopsy, and publication due to it mimicking various cutaneous neoplasms.
- Most commonly, macular amyloidosis occurs in the 4th decade, lichen amyloidosis in the 5th decade, and nodular amyloidosis in the 6th decade.
- Primary cutaneous amyloidosis affects both genders, but MA is four times more common in females.
What causes primary cutaneous amyloidosis?
The precise cause of PCA remains unclear, especially at the molecular level of amyloid fibril formation. However, lichen and macular amyloidosis are described as being of keratinocyte origin that results from apoptosis of keratinocytes with locally produced amyloid protein.
Scratching, scrubbing, and sun exposure are thought to be contributory factors. Familial tendency occurs in 10–15% of cases, and the involvement of oncostatin-M-receptor (OSMR) and IL-31 receptor A mutation has been reported.
In nodular amyloidosis, amyloid light chain (AL) deposits are produced from local plasma cell proliferation.
What are the clinical features of primary cutaneous amyloidosis?
The clinical features of primary cutaneous amyloidosis will depend on the subtype.
Lichen amyloidosis
- Characterised by 2–3 mm discrete red to dark brown (depending on individual skin type) keratotic papules,
- Commonly appear on the anterior legs and infrequently extensor surface of upper limbs, back and head.
- Some papules can coalesce forming large plaques with a cobblestone appearance.
- Rarely reported at sites such as eyelids, conchae, tongue, and anosacral areas.
- Intense itching is a prominent symptom.
- Characterised by red to dusky pigmented macules arranged in a rippled or reticulated pattern appearing over the upper back, and less frequently the chest and upper limbs.
- Pruritus in MA is less intense than LA.
Nodular amyloidosis
- Typified by amber or brown solitary or multiple nodules (5–20 mm).
- Appear over acral sites (distal limbs, head, and neck), and less commonly other parts of the body.
What are the complications of primary cutaneous amyloidosis?
Both lichen and macular amyloidosis have a benign behaviour and do not spread to other parts of the body or internal organs.
- Psychological distress can arise from skin disfigurement and itch.
- Nodular lesions have potential to progress to systemic amyloidosis in about 10% of cases.
How is primary cutaneous amyloidosis diagnosed?
The diagnosis of PCA subtypes can be made on the characteristic clinical appearance with itch at typical sites (e.g. itchy papules over shin leg for LA, and itchy hyperpigmented macules over upper back for MA).
Skin biopsy may identify amyloid deposits in the skin on H & E stained sections, Congo-red stain under polarised light illumination (apple-green birefringence), and immunohistochemistry.
Typical histologic findings show:
- Lichen amyloidosis includes large focal deposits of amyloid in dermal papillae displacing rete ridges without involvement of adnexal structures and blood vessels. Epidermal acanthosis, hyperkeratosis, papillomatosis, and hypergranulosis are seen.
- Macular amyloidosis usually exhibits small deposits of amyloid protein in the upper dermis with a relatively preserved normal epidermis.
- Nodular amyloidosis shows amyloid deposits through the entire dermis to subcutis involving skin adnexae and vessels.
What is the differential diagnosis for primary cutaneous amyloidosis?
Lichen amyloidosis especially in early or mild stage may be difficult to differentiate from other pruritic papular skin disorders, such as:
- Nodular prurigo
- Hypertrophic lichen planus
- Lichen simplex chronicus
- Pretibial myxoedema
- Pretibial pruritic papular dermatitis.
Macular amyloidosis is to be differentiated from other hyperpigmentary disorders, such as:
- Postinflammatory hyperpigmentation
- Drug-induced hyperpigmentation
- Pityriasis versicolour
- Atrophic lichen planus
- Phototoxic contact dermatitis
- Erythema dyschromicum perstans
- Notalgia paresthetica (amyloid may develop however due to scratching)
- Poikiloderma of Civatte.
Nodular amyloidosis can mimic various cutaneous and subcutaneous nodules, including:
- Sarcoidosis
- Cysts
- Neoplasia, including basal cell carcinoma
- Mycosis fungoides
- Cutaneous pseudolymphoma
- Colloid milium
- Leiomyoma.
What is the treatment for primary cutaneous amyloidosis?
Due to the lack of controlled trials, there are no accepted treatment strategies. With all therapies, there is recalcitrance and recurrences. Treatment focuses on relief of aesthetic disfigurement and pruritus.
General measures
- Moisturiser and emollient use
- Avoidance of scratching
Specific treatment
Topical treatment
- Corticosteroids: Potent or super-potent topical corticosteroids are widely used and are more effective under occlusion.
- They can be used with keratolytic agents such as urea, salicylate acid, or lactic acid, improving elimination of the keratosis, lichenification, and flattening of papules.
- Calcineurin inhibitors
- Retinoids
- Dimethyl sulfoxide (DMSO 10%), and menthol 2%
- Antipruritic medications such as capsaicin or doxepin may be trialled, but its supportive evidence is limited
- Use of nocturnal sedating antihistamines seemed to minimise scratching rather than pruritus
- Intralesional corticosteroid injection or cryosurgery with or without combined with topical steroids depending on the extent of the lesion and tolerance.
Systemic/oral treatment
Useful for extensive lesions when topical treatments are impractical or unresponsive — (used off-label):
- Oral retinoids (e.g. acitretin, alitretinoin)
- Antineoplastic and immune modulators (e.g. cyclophosphamide, ciclosporin, and dupilumab) have been advocated, but their use would be only in exceptional circumstances.
Physical treatment
- Phototherapy: PUVA and narrow-band UVB
- Laser-therapy: Fractional CO2, Er:YAG, Nd:YAG, and Pulse Dye Laser
- Dermabrasion, cautery and curettage, or excision (depending on location and extent of the lesion).
What is the outcome of primary cutaneous amyloidosis?
Primary cutaneous amyloidosis is often resistant to therapy, and if it does improve, it is likely to recur when any therapy is discontinued.