Introduction
Demographics
Causes
Clinical features
Variation in skin types
Complications
Diagnosis
Differential diagnoses
Treatment
Outcome
Mycosis fungoides is the commonest type of primary cutaneous T-cell lymphoma, a form of non-Hodgkin lymphoma characterised clinically by progression from patches to plaques to tumours and, on histology, by an epidermotropic infiltrate of small to medium-sized CD4+ T-cells (lymphocytes).
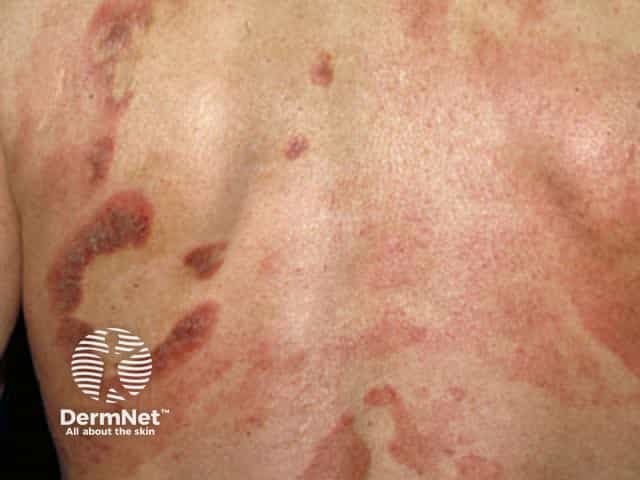
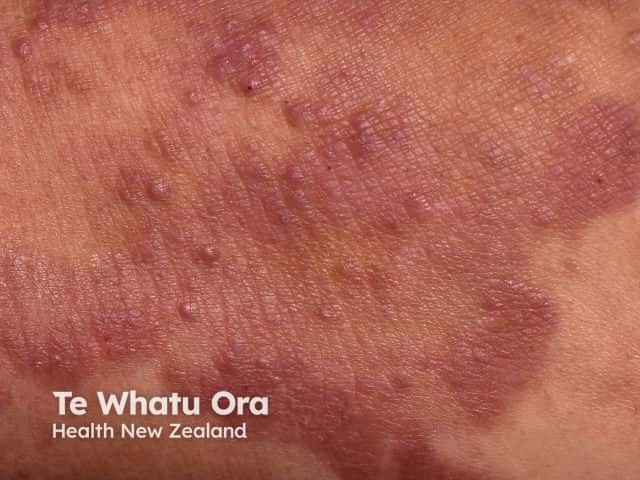
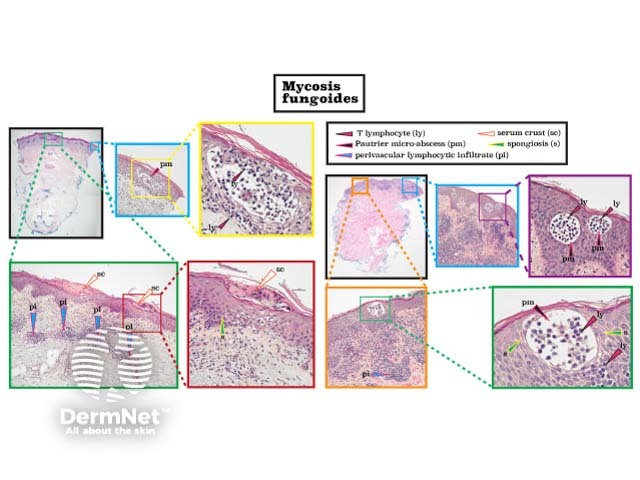
Mycosis fungoides (MF) represents 50% of all primary cutaneous lymphomas and 60-70% of all cutaneous T-cell lymphomas (CTCL). However, it is an uncommon condition with an estimated incidence worldwide of 6.4 per million.
Onset is usually in late adulthood (median age 55–60 years) with a male predominance (2:1) in white patients. Diagnosis in patients under 30 years of age is highest in Asians, Pacific Islanders, and Black Americans.
The aetiopathogenesis of mycosis fungoides remains unknown. Theories include chronic antigen stimulation, a genetic predisposition (eg, HLA type), or chemical exposure (eg, medications). Geographical clustering of cases and MF affecting family members suggests environmental triggers. Unlike some forms of CTCL, there is no evidence currently for a viral aetiology.
Mycosis fungoides can present as a chronic itch, even before there are clinical signs of disease. Classic MF progresses slowly over years from patches to plaques and may eventually form tumours. Most patients are diagnosed with early-stage disease.
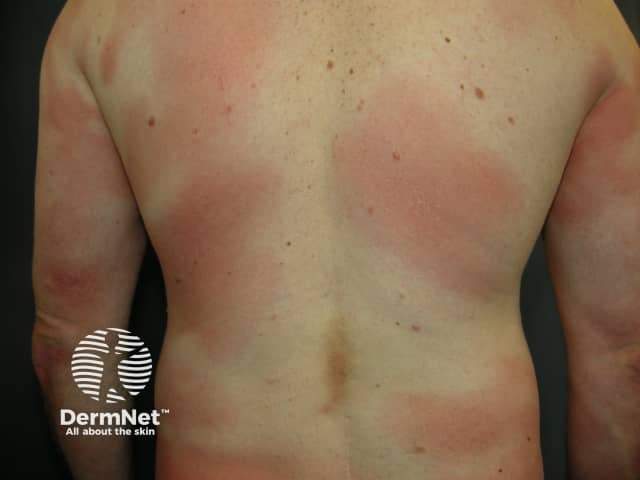
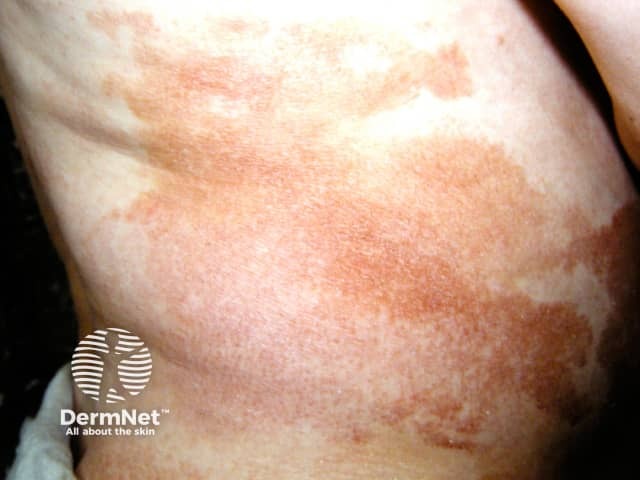
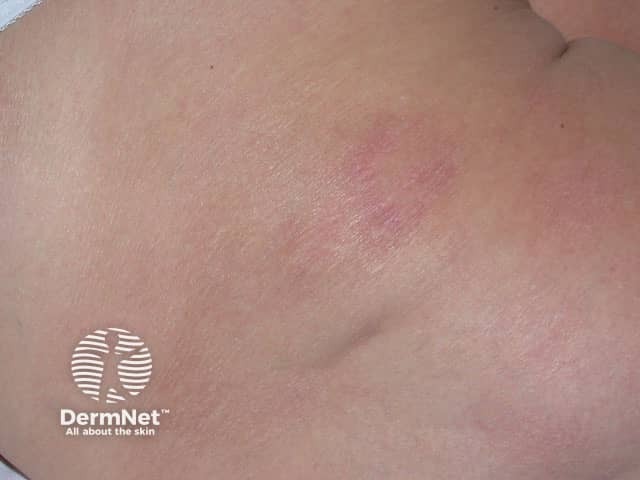
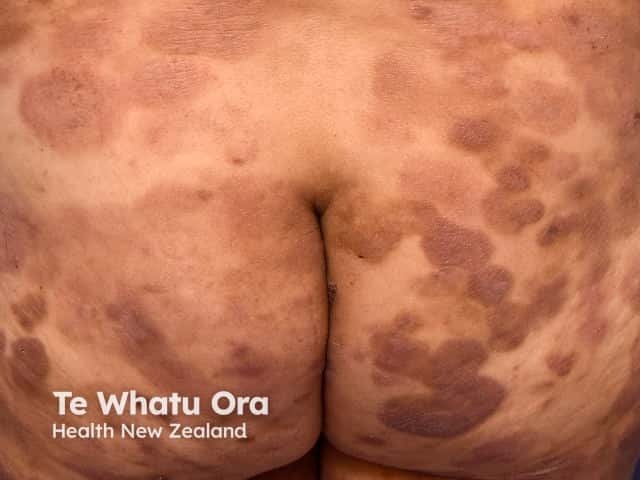
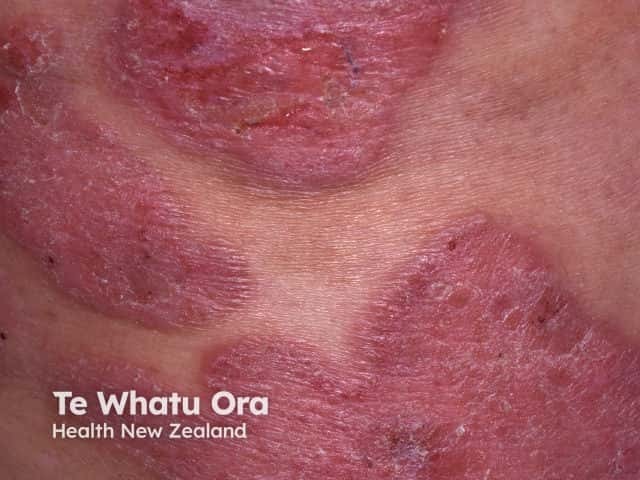
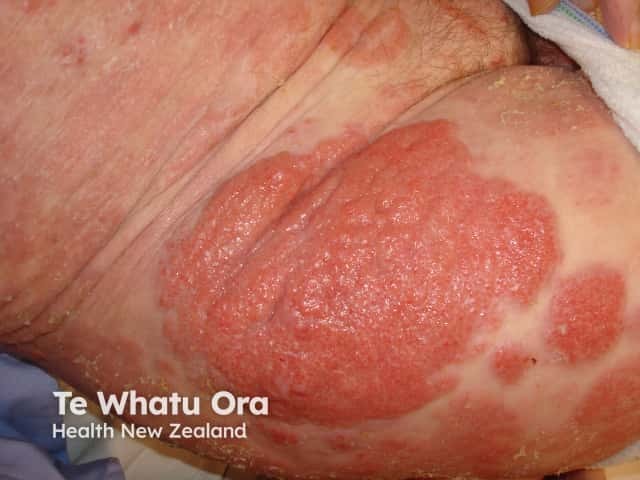
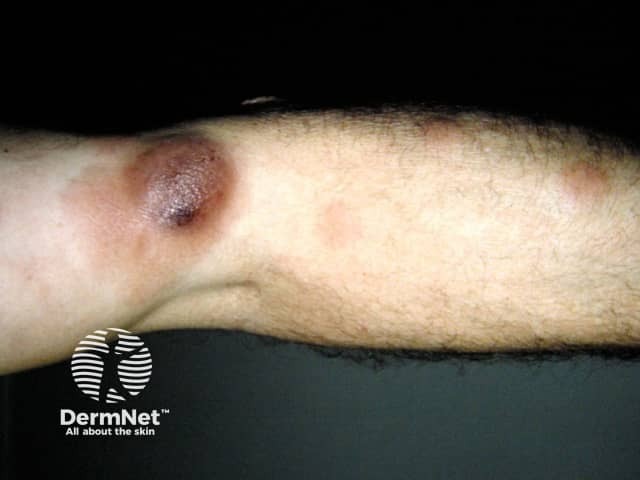
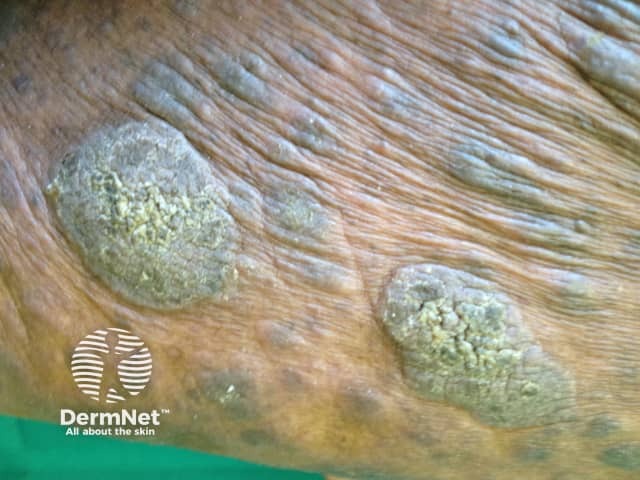
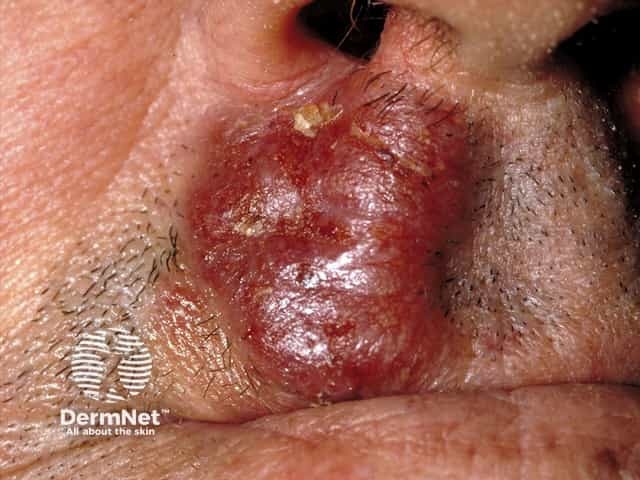
There are many clinical presentations that do not look like classic mycosis fungoides but follow the same natural history including:
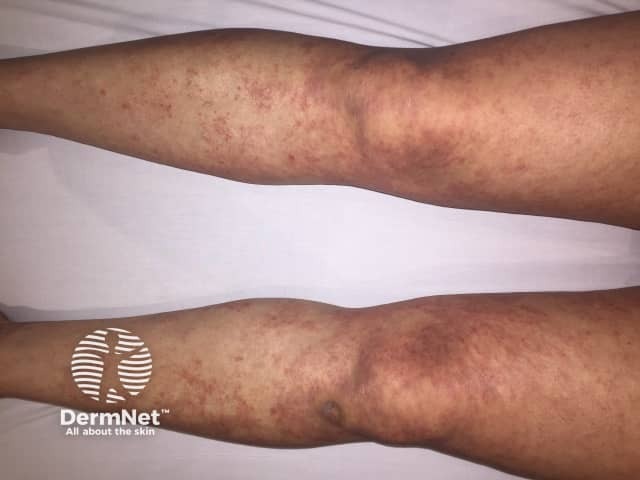
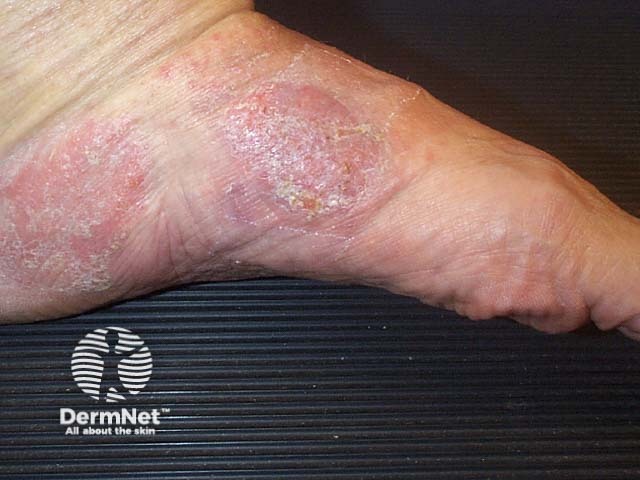
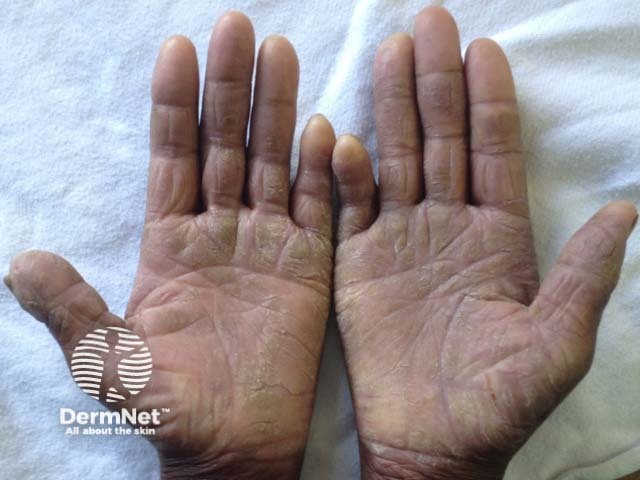
Mycosis fungoides is the most common primary cutaneous lymphoma (65%) in children but diagnosis is often delayed (2–5 years). The mean age at diagnosis is 10 years, and the sexes are affected almost equally. Most children present with non-classic and often multiple variants. More than 50% of children present with the hypopigmented variant of MF; classic MF affects 40%, and folliculotropic MF in one-third.
Mycosis fungoides in skin of colour often presents as hypopigmented papules and plaques without epidermal atrophy. A large case series from Johns Hopkins reported Black American males presented with later stage disease and greater skin involvement than white Americans; Black American females had early-onset aggressive disease. Patients with hypopigmented lesions were more likely to have early-stage disease and better survival than those with advanced-stage disease in another US series.
The diagnosis of mycosis fungoides requires careful clinicopathological correlation. Dermoscopy can help to distinguish MF from inflammatory dermatosis, other causes of erythroderma, and leprosy (see Dermoscopy of leprosy).
Multiple skin biopsies are often required over time to confirm the diagnosis especially in the early stages. Histopathology is characterised by infiltrates of malignant CD4+ T-cells (see Mycosis fungoides pathology).
Further investigations performed on skin biopsy specimens may include:
Blood tests, lymph node biopsy, and imaging (eg, CT/PET scans) may be required if there is concern the skin lesions are secondary to a systemic lymphoma, in advanced disease (ie, extracutaneous spread), or for TNMB staging.
Skin staging of mycosis fungoides: T1, limited (<10% BSA) patches/plaques; T2, generalised (>10% BSA) patches/plaques; T3, tumours; T4, erythroderma.
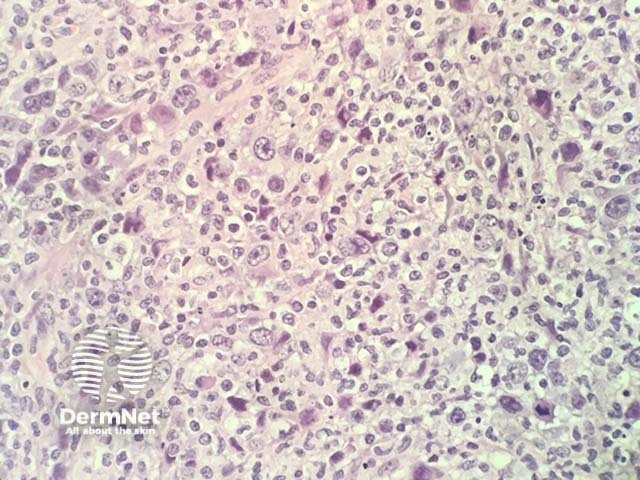
Mycosis fungoides is treatable, not curable. There are no globally accepted treatment guidelines. Treatment should be guided by the patient’s wishes, stage of disease, availability of options, and local expertise. Disease relapse is common after most treatments. A wait-and-see approach can be considered for early limited disease.
Only brentuximab vedotin (for CD30+ MF) and allogeneic haematopoietic stem cell transplant alter the natural history of the disease.
Mycosis fungoides typically follows an indolent course over years, with an estimated 5-year survival of 87%. The duration of early-stage disease has been estimated to be approximately 18 years. Although improved survival with MF has been demonstrated, it is likely due to earlier diagnosis rather than advances in therapy.
Individual lesions can sometimes remit spontaneously.
Age, skin stage, and the presence/absence of extracutaneous disease are the major predictors of survival. Skin stage T1 disease (limited patches/plaques) does not alter life-expectancy, although approximately one-third of patients with skin stage T1 disease progress to more advanced stages.
Hypopigmented and poikilodermatous variants have reduced disease progression and better disease-specific survival compared to classic MF while folliculotropic, pustular, and granulomatous MF are considered to be more aggressive variants.
The most common cause of death is non-Hodgkin lymphoma (NHL), which is usually the mycosis fungoides but can be a second NHL, with non-NHL malignancy being the second most common.

