Introduction Demographics Causes Clinical features Complications Diagnosis Differential diagnoses Treatment Outcome
Bullous systemic lupus erythematosus (SLE) is an autoimmune subepidermal blistering disease that occurs in patients with SLE. It can be associated with antibodies against type VII collagen.
Bullous SLE is also called bullous eruption of SLE and vesiculobullous SLE.
The incidence of bullous SLE was estimated to be 0.22 and 0.26 cases per million per year in France and Singapore. In a large cohort of sera taken from patients with immunobullous disorders, 1–2% were identified as bullous SLE [1,2].
Like SLE, bullous SLE has been reported most commonly in women of African descent in their thirties. However, bullous SLE occurs in all ages, sexes, and ethnicities.
Bullous SLE may be the presenting sign of SLE in approximately one third of patients. In the majority though, SLE has already been diagnosed, in some cases many years prior.
Bullous SLE is classified into three types based on immunohistochemistry.
The three types cannot be distinguished clinically.
Predisposing factors include adverse drug reactions to hydralazine, penicillamine, methimazole; and exposure to ultraviolet (UV) B radiation.
Bullous SLE type I is similar to epidermolysis bullous acquista (EBA) in that both have autoantibodies to type VII collagen on indirect immunofluoresence [6]. However bullous SLE and EBA differ clinically and the IgG autoantibodies have now been shown to be different.
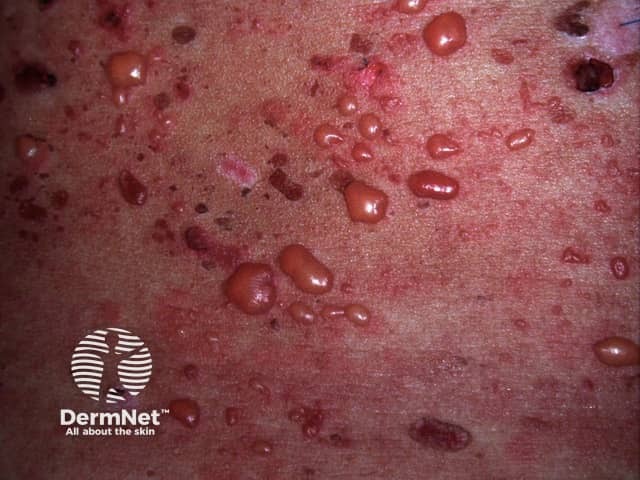
In bullous SLE, tense vesicles, bullae and erosions arise on normal or erythematous skin, usually in sun-exposed sites.
The complications of bullous SLE are those of the underlying SLE. The development of bullous SLE is often associated with a flare of lupus nephritis.
Investigations are necessary to distinguish bullous SLE from SLE with a concomitant blistering disorder.
Histopathological findings resemble dermatitis herpetiformis, in that they both show:
Basal layer vacuolation, characteristic for other forms of cutaneous LE, is not present.
The characteristic direct immunofluorescence (DIF) finding in a perilesional skin biopsy of bullous SLE is linear or granular immunoglobulin (Ig)G, IgM, IgA or C3 at the dermoepidermal junction (DEJ). Type I bullous SLE is typically associated with the linear pattern.
Serum autoantibodies detected by the indirect immunofluorescence test or enzyme-linked immunosorbent assay (ELISA) are against type VII collagen in type I bullous SLE. BP 230, BP 180, or laminin 5 or 6 are detected in type II bullous SLE.
Serration pattern analysis of DIF microscopy differentiates tissue bound auto-antibodies against type VII collagen, where a u-serrated pattern is seen, from all other anti-DEJ antibodies where an n-serrated pattern is seen [7].
Diagnostic criteria for bullous SLE requires:
The differential diagnosis of bullous SLE is SLE with blisters. Other blistering conditions that may arise in SLE include:
Dapsone, at a dose of 1.0–1.5 mg/kg/day, is considered the treatment of choice. It is effective in the vast majority of patients and leads to rapid clinical improvement within days or a few weeks. This rapid response differentiates bullous SLE from SLE with a concomitant autoimmune blistering disease.
Where dapsone fails, prednisolone (systemic steroids) and azathioprine are used. Patients successfully treated with methotrexate or rituximab have been reported [8–11].
Bullous SLE is transient in the majority of cases and usually completely regresses with no further flares, irrespective of the activity of the systemic disease. It typically resolves without milia or scarring but can leave hypo- or hyper-pigmentation.

