Introduction
Approach to a suspected drug reaction
Diagnosis
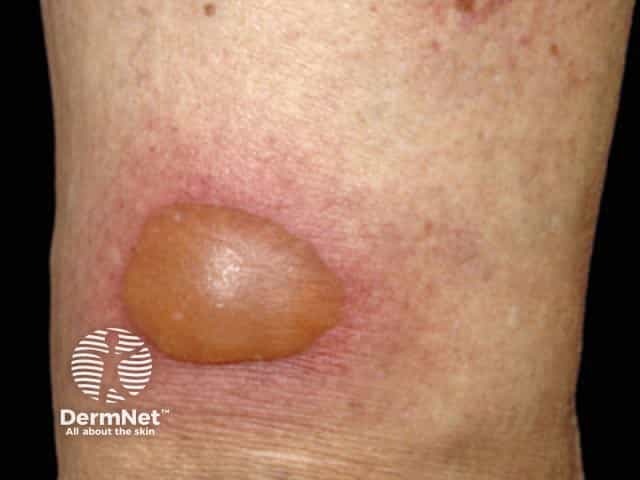
Mild blistering reaction in a restricted area
Approach to the acute generalised blistering patient
The term bullous drug eruptions refers to adverse drug reactions that result in fluid-filled blisters or bullae.
Blistering can be due to various medications, prescribed or over-the-counter, natural or synthetic. Blistering may be localised and mild, or widespread and severe, even life-threatening. Blisters may be the major feature of the reaction or may be only seen sometimes or in localised areas of a more extensive rash. The reaction may show features of more than one condition (overlap) or be currently unclassifiable.
It is important to tell the doctor about all medications, even those purchased without a prescription and including ‘natural’ or herbal remedies, as ceasing the causative agent is the single most important part of the treatment to limit the damage.
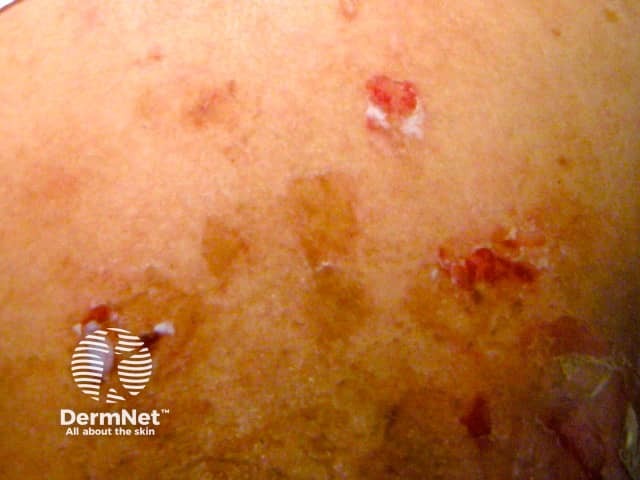
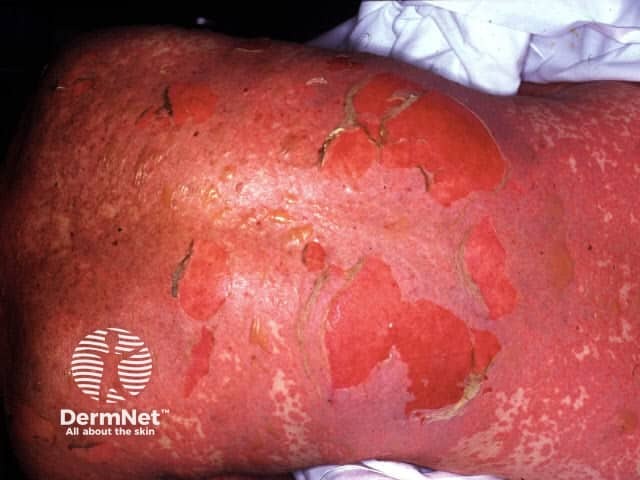
When a drug reaction is suspected, the presence of blisters would almost always necessitate ceasing the likely drug.
Bullous drug eruptions are nearly always diagnosed clinically, (ie, by careful history and examination).
A skin biopsy may be required to make the correct diagnosis,but does not usually help in establishing whether the reaction is drug-induced. Unfortunately, apart from rechallenge with the drug, there are usually no reliable tests to determine if the suspected drug has caused the rash. Skin tests (eg, patch test, intradermal injection or pinprick), in vitro lymphocyte transformation test and in vitro drug-induced interferon gamma test can sometimes be helpful in confirming the role of specific drugs in a reaction. Rechallenge is not recommended after a serious reaction.
Fixed drug eruption (FDE) always recurs at the same site(s) on re-exposure to the drug/agent. The most common sites are the lips, genital area, hands and feet. On the first occasion, it begins 1–2 weeks after drug exposure as one or more sharply defined red swollen lesions that may develop a central clear blister. It settles over several days, leaving round to oval shaped area(s) of flat pigmentation. With every further exposure, the reaction appears more quickly (ie, within 24 hours), is more inflamed, and the residual pigmentation darkens. Sometimes the number of sites also increases with each re-exposure.
Antibiotics are the commonest cause, especially sulphonamides and tetracyclines, but many other drugs have been reported including terbinafine and anti-inflammatory drugs (NSAIDs).
Provocation with the possible agent (after allowing for a possible refractory period) will confirm the diagnosis. Patch testing with the agent at the site of the fixed drug eruption, but not elsewhere on the skin, can elicit a positive response.
The mechanism of this reaction involves immune cells (lymphocytes) called memory CD8+ T cells that remain in a fixed area of skin.
Pseudoporphyria is defined as skin changes consistent with one of the porphyrias, but laboratory tests do not detect significant abnormalities in the porphyrin-haem pathway.
Erythropoetic protoporphyria (EPP)-like pseudoporphyria is usually seen in children with juvenile rheumatoid arthritis taking naproxen. It presents with skin fragility, scarring and vesicles (small blisters) in sun-exposed sites, mainly the face (nose, cheeks). In one large study of children taking naproxen for juvenile idiopathic arthritis, signs of pseudoporphyria appeared on average 18 months after starting the drug and usually within the first two years. Onset was not seasonal. The risk of children developing pseudoporphyria when taking naproxen was 11%. Identified risk factors included:
The use of sunscreens did not appear to prevent the development of this pseudoporphyria.
A porphyria cutanea tarda (PCT)-like form seen in adults presents with tense blisters (95%) photosensitivity (sun sensitivity; 65%), skin fragility (65%), scarring (65%) and milia (‘whiteheads’; 40%), in sun-exposed sites including the backs of hands and face. Other clinical features of true PCT are not seen.
Skin biopsy shows changes indistinguishable from true porphyria cutanea tarda. However urine, faecal and blood porphyrins are normal. It is therefore a diagnosis of exclusion.
The most commonly reported drug to cause PCT-like pseudoporphyria is also naproxen and other related propionic acid derived non-steroidal anti-inflammatory drugs (NSAIDs). Sulindac, indomethacin and diclofenac appear to be safe alternative NSAIDs.
The skin changes usually improve 1–6 months after ceasing the drug, but may persist indefinitely. New lesions may continue to appear weeks or months after ceasing naproxen. Facial scarring improves slowly with time. Many other medications have been reported to cause pseudoporphyria including antibiotics, diuretics and retinoids, such as acitretin and isotretinoin.
Pseudoporphyria is probably a form of non-immune-mediated drug photosensitivity.
It is important to remember that true PCT can be precipitated by oestrogen (oral contraceptive pill, hormone replacement therapy) and alcohol.
There are two mechanisms of drug-induced photosensitivity — phototoxic and photoallergic reactions. Photoallergic reactions do not blister.
Phototoxicity presents as an exaggerated sunburn reaction; that is, a sunburn that has occurred at less than the usual sun exposure required, or a more severe sunburn reaction than would usually have occurred for that amount of sun. This may include a blistering sunburn reaction. It occurs where the skin has been exposed to the sun and so typically involves face, neck and upper chest.
The most common drugs to cause this reaction are:
It is sometimes possible to continue the drug by reducing sun exposure, reducing the drug dose or taking the drug at night if it has a short half life in the body.
A phototoxic reaction is predictable and requires only a sufficient drug dose and sufficient ultraviolet exposure (sun, solarium) to occur. It involves a direct interaction between ultraviolet light and the drug (or its breakdown product).
Hand foot syndrome is a common reaction to chemotherapy drugs and occurs on palms and soles. It begins as painful redness which then becomes puffy and swollen. The colour darkens and blisters may develop. It usually develops within days of the first treatment but onset can be delayed up to 10 months.
The first warning is often tingling of the palms and soles before the symmetrical, sharply defined redness appears. Palms are affected more commonly than soles. Initially, it settles quickly 1–5 weeks after the drug is ceased, but recurs more severely with each cycle and takes longer to resolve. It becomes more common with each chemotherapy cycles. The skin changes disappear without scarring unless there has been ulceration.
Many chemotherapy agents have been reported to cause hand-foot syndrome, however, the commonest is 5-fluorouracil infusion and its oral prodrug capecitabine, (because of the frequency of use) but pegylated liposomal doxorubicin is the commonest per dose (50% of patients treated with the recommended dose).
The only treatment shown to definitely have any benefit is to cease therapy or reduce the drug dose or frequency. Although it is not life threatening, it affects the quality of life. Regular use of moisturisers and avoiding activities that may cause heat, pressure, friction or irritation of the skin are recommended.
It appears the chemotherapy drug reaches the skin surface in sweat where it accumulates and then has a direct cytotoxic effect on the skin cells.
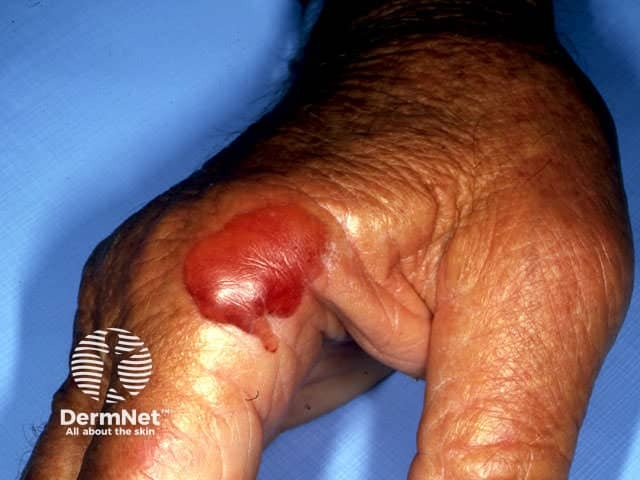
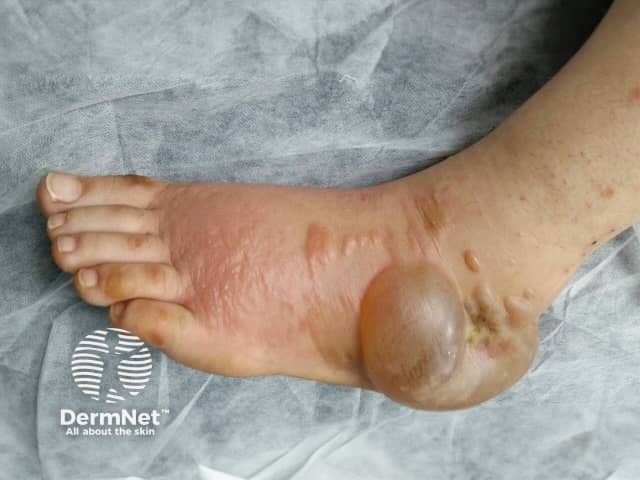
Any medication that can cause peripheral oedema (puffiness and swelling of feet and lower legs) can result in blistering localised to the swollen areas. Typically the blisters are tense and filled with clear fluid, developing in puffy swollen skin on the tops of the feet and ankles. The blisters develop slowly and may reach several cm in diameter. Calcium channel blockers (used to treat high blood pressure) are the most common drug cause of this.
Although this eruption is usually acne-like with pustules, it can be bullous with either clear or blood-filled blisters (especially if due to iodine). Usually, it develops after long term exposure to halogens (topical or oral iodine, oral bromide) and resolves slowly over weeks after ceasing the drug due to slow excretion through the kidneys. Acute or chronic renal failure is a risk factor, particularly for acute halogenoderma development such as after oral or intravenous iodine-containing radiocontrast medium for X-rays. Treatment may involve the use of intravenous fluids or oral fluid tablets to increase halogen excretion, topical or systemic corticosteroids and, if severe, ciclosporin. Formation of halogenoderma seems to involve the accumulation of the halogen as well as a hypersensitivity/allergy.
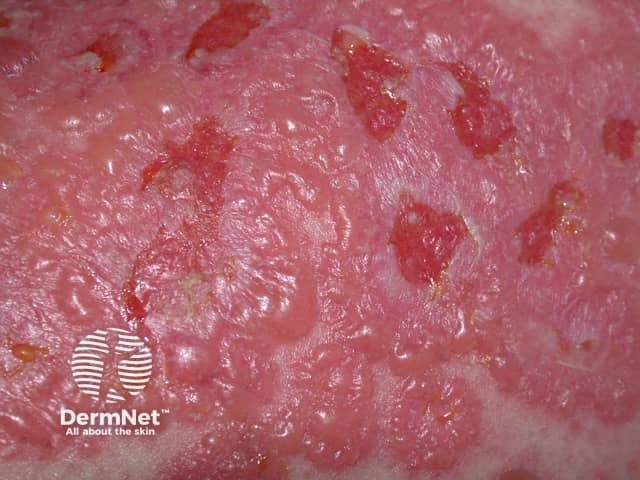
Cutaneous small vessel vasculitis usually presents as palpable purpura (ie, a slightly raised non-blanching red-purple spotty eruption usually of the lower limbs that may also develop pustules and blood-filled blisters). Approximately 10% of cases are due to drugs, developing 1–3 weeks after drug administration.
The blisters, if present, are small and develop on top of the spots. There may be associated symptoms such as fever, muscle and joint pain and headache.
Skin biopsy of an early lesion is recommended as confirmation of the diagnosis necessitates further investigations.
The most common drug causes of vasculitis are:
Cessation of the causative drug results in rapid improvement. However, rechallenge will result in recurrence of the rash within 3 days.
Leukocytoclastic vasculitis is the histopathological term for hypersensitivity vasculitis that presents as palpable purpura. It is caused by the deposition in small blood vessels of immune complexes of antibodies and drug-related allergens. These immune complexes then activate inflammatory pathways that result in leaky blood vessels.
1. History
2. General examination
3. Blisters
4. Histopathology
4. Further investigations
Clinically drug-induced bullous pemphigoid usually resembles classic bullous pemphigoid with urticaria (hive)-like patches and tense clear blisters that do not break easily. They usually appear suddenly. Sometimes it can look more like erythema multiforme. It rarely affects the mucous membranes. Generally, it occurs in a younger age group than classic idiopathic bullous pemphigoid, which is a disease of old age.
The skin biopsy, histopathology, and direct immunofluoresence is the same as for the classic disease.
Drugs reported causing this reaction include:
Prompt cessation of the drug results in rapid clearance of the rash. Although the exact mechanism is not known, there may be an underlying genetic susceptibility that modifies the immune response to the drug or alters the antigenic properties of the epidermal basement membrane.
In developed countries, approximately 10% of cases of pemphigus are drug-induced or drug-triggered.
The skin lesions are flaccid blisters which break easily and often only erosions +/− crusting are seen. The Nikolsky sign can be positive. In drug-induced pemphigus, mucous membranes are only involved in 10–15% of cases whereas drug-triggered pemphigus is indistinguishable from classic pemphigus.
Skin biopsy shows the typical separation of individual skin cells seen in pemphigus. Direct immunofluoresence (DIF) is positive in 90% of drug-related pemphigus, compared to 100% in idiopathic classic disease. Circulating anti-desmoglein autoantibodies are only detected in the blood in 70% of drug-induced pemphigus.
The onset of drug-related pemphigus can be weeks to months after the drug was started. Resolution occurs after drug withdrawal in drug-induced pemphigus but not if drug-triggered.
Drug-induced pemphigus is caused by drugs with a thiol group, such as:
It is believed the thiol group binds to a desmosomal antigen and the complex stimulates an immune reaction against the desmosome. Desmosomes hold the skin cells together and when these are damaged the skin cells separate resulting in a blister. However, drugs in this group when tested in the laboratory can cause skin cells to separate suggesting a direct effect on the epidermis without an immune response.
Drug-triggered pemphigus follows non-thiol drug use including:
Linear IgA bullous dermatosis can be idiopathic or associated with systemic diseases or drug exposure. The drug related form usually resembles the idiopathic type with tense small and large blisters often in ring-shaped arrangements on the body, arms and legs. It may involve the palms and soles and rarely mucous membranes. Rarely it may be more severe and resemble toxic epidermal necrolysis.
Skin biopsy shows the same histopathology and direct immunofluoresence (DIF) features as in the idiopathic form. The DIF becomes negative after the drug has been withdrawn and the rash settles.
On blood testing, the drug-related form differs from the idiopathic type in that there is usually no detected circulating IgA autoantibody to the basement membrane zone. If it is detected, then it too disappears with the resolution of the rash.
The reaction begins 1–2 weeks after starting the drug (range 24 hours to 15 days). When the offending drug is ceased, new blisters stop appearing 1–3 days later and the rash has usually cleared by 3 weeks. Rechallenge with the drug results in a more severe reaction with a shorter latency time and longer time to clearance.
The most common drug associated with this reaction is vancomycin; however, other drugs have also been reported to cause it.
The suggested mechanism is that the drug activates immune cells (lymphoctyes) called CD8+ T cells, which release Interleukin (IL)-5. This cytokine is known to influence IgA expression. The target antigens for the IgA antibody are numerous and the same as for idiopathic linear IgA bullous dermatosis.
This reaction was formerly called ‘hypersensitivity syndrome’ or ‘drug-induced hypersensitivity syndrome’. It is defined as the triad of fever, skin eruption and internal organ involvement. It is always caused by a drug.
Fever and rash are the commonest signs. The rash begins as a patchy or diffuse redness (morbilliform) and progresses to small and large tense blisters because of skin swelling (oedema). Swelling of the face is a particular feature. The rash then spreads to involve the body, arms and legs. Mucous membranes are not affected. Lymph node enlargement is common. The commonest internal organ involved is the liver (50%), but kidneys, lungs, heart, GIT and thyroid can also be affected.
Skin biopsy shows a dense infiltrate of specific white cells (lymphocytes and eosinophils) in the upper dermis with swelling. The lymphocytes can look atypical, resembling lymphoma (pseudolymphoma). DIF is negative.
There is a very high eosinophil cell count in the blood (eosinophilia). Other tests are usually required for the liver, kidneys, etc.
This reaction usually begins 2–6 weeks after the drug was started. It is probably more common in patients of African descent. The most common cause of DRESS is aromatic antiepileptic medication (phenytoin, carbamazepine, phenobarbital), with a risk of 1/5000. Cross-reactivity between these drugs is well documented.
Stopping the drug early is important but may not result in full recovery. Treatment, which may be required for weeks or months, may include either topical high potency topical corticosteroids (if the reaction is mild and mainly involving the skin) or systemic corticosteroids for lung and heart involvement. The fever, rash and hepatitis may persist for several weeks or longer. Carbamazepine and allopurinol reactions have been reported to last as long as 10 and 16 months respectively. The mortality rate is said to be about 10%, mainly due to severe hepatitis. Follow-up, often required for several months, is important especially for the liver, kidney and thyroid function and monitoring of the full blood count for the eosinophil count.
The mechanism for this reaction may involve a number of factors including:
Stevens–Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN) are now regarded as clinical variants of the same condition and are caused by medications in most if not all cases.
SJS/TEN begins with 2–3 days of fever often in association with sore eyes (conjunctivitis), sore throat (pharyngitis) and itch. Mucous membrane (mouth/throat, eyes, genital area and anus) involvement often precedes the development of the skin changes by 1–3 days. Painful flat red patches with darker centres appear, starting on the face and upper body, then spreading to the rest of the body and limbs. Over hours to days flaccid (thin-walled) blisters and sheets of surface skin loss develop. The blisters extend with skin pressure and the Nikolsky sign is positive. Where the skin is lost, there is redness and oozing.
Blood tests are virtually always abnormal. A skin biopsy may be done to distinguish this from staphylococcal scalded skin syndrome. It shows the death of the full thickness of epidermis with little inflammation. DIF is negative.
SJS/TEN results in a very sick patient and the mortality rate can be as high as 30%. The SCORAD assessment provides an estimate of severity and mortality. It is therefore important to recognise early, to cease the offending drug and admit to hospital in a burns intensive care unit if possible. A multidisciplinary team approach is required. Care is mainly supportive, pre-empting possible complications and should include pain relief, fluid and nutritional replacement, prevention of infection, maintaining body temperature, mouth, skin and eye care. Blood clots should be prevented with heparin and the patient should be frequently re-assessed for the involvement of the respiratory tract, gastrointestinal tract and urinary tract. Management of the associated anxiety is important. The use of systemic corticosteroid treatment is debated. IVIG (intravenous immunoglobulins) and ciclosporin have been reported to be of some benefit although this also is not consistent.
More than 100 drugs have been reported to cause SJS/TEN. The most commonly implicated medications are sulphonamides (and other antibiotics) and anti-epilepsy medications. The onset varies from a few days up to one month except for anti-epilepsy drugs where it may be as long as 8 weeks after starting.
The mechanism is probably immunological involving T lymphocytes and Fas-Fas ligand-induced keratinocyte apoptosis.
See Steven Johnson syndrome / toxic epidermal necrolysis: nursing management
This is a rare, sometimes fatal, reaction to either warfarin or heparin via different mechanisms.
Warfarin necrosis begins 2–5 days after starting the drug initially as pain and redness over the breasts, thighs and/or buttocks. This progresses to blood-filled blisters then black, necrotic ulcers. Treatment is to stop the warfarin replacing it with heparin, giving vitamin K supplements and intravenous protein C infusions. It affects 1/10,000 patients started on warfarin and is more common in those with an inherited deficiency of protein C. Other risk factors are a high initial warfarin dose, obesity and female sex. It is due to a fall in protein C function resulting in blood clots in the blood vessels of the skin and subcutaneous fat. The clots block the blood supply to the skin, resulting in skin death.
Heparin necrosis begins later (average 7 days after starting heparin) as redness and pain, then blisters and skin necrosis usually at the heparin injection sites but sometimes also elsewhere on the skin. There are a number of different mechanisms, but the commonest is an immune system reaction to a heparin-induced change in the platelet factor 4 complex, resulting in a fall in platelet count and formation of clots. None of the heparin necrosis mechanisms involve Protein C or Protein S. Treatment is to stop the heparin, and replace with other anti-coagulant drugs if required.
Pain relief and wound care of areas of skin necrosis and sometimes surgery may also be required.
Other uncommon drug causes of generalised blisters include:

